Featured Publications

All Publications
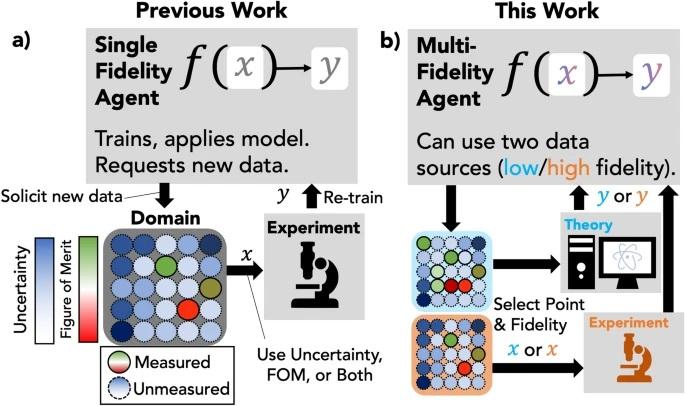
Sequential learning for materials discovery is a paradigm where a computational agent solicits new data to simultaneously update a model in service of exploration (finding the largest number of materials that meet some criteria) or exploitation (finding materials with an ideal figure of merit). In real-world discovery campaigns, new data acquisition may be costly and an optimal strategy may involve using and acquiring data with different levels of fidelity, such as first-principles calculation to supplement an experiment. In this work, we introduce agents which can operate on multiple data fidelities, and benchmark their performance on an emulated discovery campaign to find materials with desired band gap values. The fidelities of data come from the results of DFT calculations as low fidelity and experimental results as high fidelity. We demonstrate performance gains of agents which incorporate multi-fidelity data in two contexts: either using a large body of low fidelity data as a prior knowledge base or acquiring low fidelity data in-tandem with experimental data. This advance provides a tool that enables materials scientists to test various acquisition and model hyperparameters to maximize the discovery rate of their own multi-fidelity sequential learning campaigns for materials discovery. This may also serve as a reference point for those who are interested in practical strategies that can be used when multiple data sources are available for active or sequential learning campaigns. READ MORE
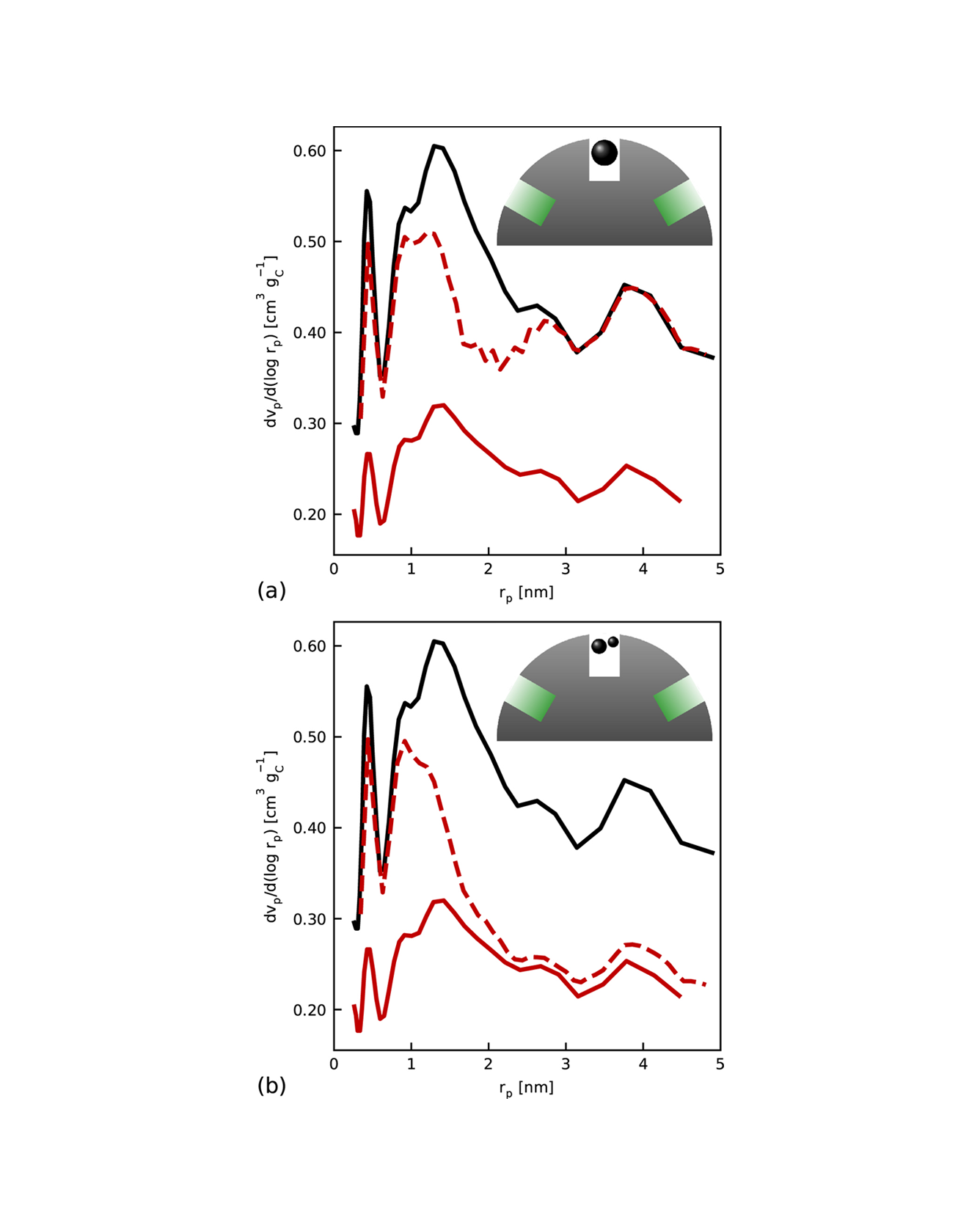
We present a model of the cathode catalyst layer morphology before and after loading a porous catalyst support with Pt and ionomer. Support nanopores and catalyst particles within pores and on the support surface are described by size distributions, allowing for qualitative processes during the addition of a material phase to be dependent on the observed pore and particle size. A particular focus is put on the interplay of pore impregnation and blockage due to ionomer loading and the consequences for the Pt/ionomer interface, ionomer film thickness and protonic binding of particles within pores. We used the model to emulate six catalyst/support combinations from literature with different porosity, surface area and pore size distributions of the support as well as varying particle size distributions and ionomer/carbon ratios. Besides providing qualitatively and quantitatively accurate predictions, the model is able to explain why the protonically active catalyst surface area has been reported to not increase monotonically with ionomer addition for some supports, but rather decrease again when the optimum ionomer content is exceeded. The proposed model constitutes a fast translation from manufacturing parameters to catalyst layer morphology which can be incorporated into existing performance and degradation models in a straightforward way. READ MORE
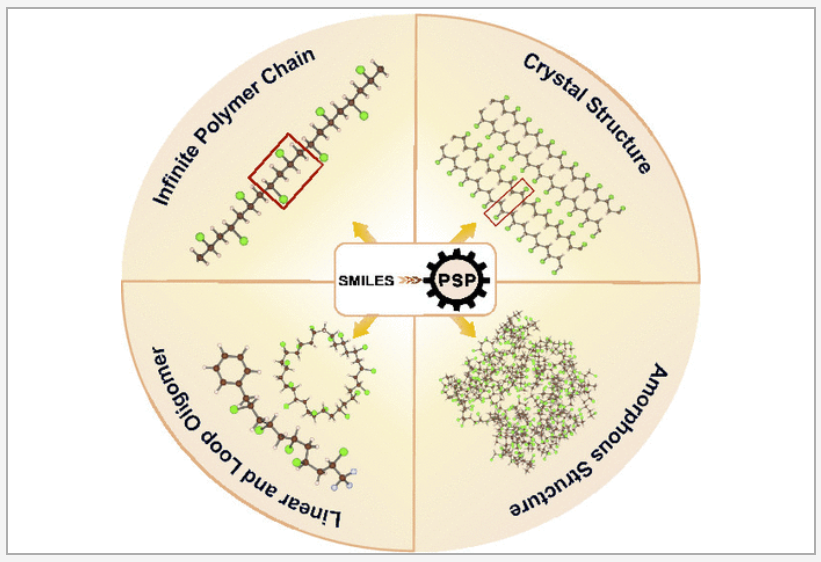
Three-dimensional atomic-level models of polymers are the starting points for physics-based simulation studies. A capability to generate reasonable initial structural models is highly desired for this purpose. We have developed a python toolkit, namely, polymer structure predictor (PSP), to generate a hierarchy of polymer models, ranging from oligomers to infinite chains to crystals to amorphous models, using a simplified molecular-input line-entry system (SMILES) string of the polymer repeat unit as the primary input. This toolkit allows users to tune several parameters to manage the quality and scale of models and computational cost. The output structures and accompanying force field (GAFF2/OPLS-AA) parameter files can be used for downstream ab initio and molecular dynamics simulations. The PSP package includes a Colab notebook where users can go through several examples, building their own models, visualizing them, and downloading them for later use. The PSP toolkit, being a first of its kind, will facilitate automation in polymer property prediction and design. READ MORE
Chapter 3
The materials discovery process naturally presents a slew of questions about the character of a material. These range from simply trying to learn basic facts about the atomic structure of the compound [1–3] all the way to producing a time-resolved profile of a functional process in operando from start to finish [4]. Spectros-copy is the process of measuring a materials’ response to external electromagnetic stimulus to deduce the properties of interest. Spectroscopy can help to solve problems in experimental design and decision-making (probing response of certain energy domains, measuring particular properties), inference (moving from raw data to the property of interest), and analysis (rationalizing and interpreting the data). These are all classes of problems which artificial intelligence (AI) is well-posed to address.As such, the interface between practitioners in both spectroscopy and AI has sparked a great deal of excitement and research activity. Because characterization is a crucial process of the materials discovery pipeline, insightful pairings of algorithms and experimental protocols can lead to gains in both experimental efficiency and accuracy.These gains may significantly compress the timescales of experimental procedures and help experimentalists learn more in less time or answer questions which were previously inaccessible with a given experimental apparatus. READ MORE

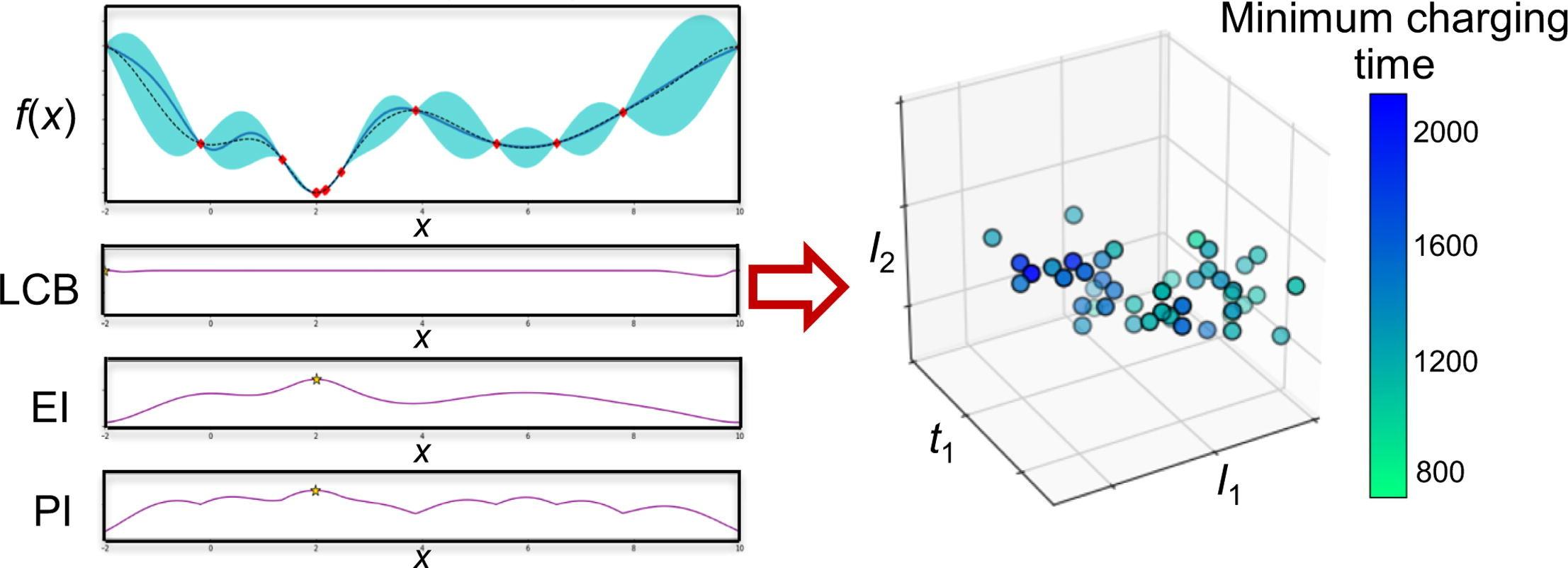
Lithium-ion batteries are one of the most commonly used energy storage device for electric vehicles. As battery chemistries continue to advance, an important question concerns how to efficiently determine charging protocols that best balance the desire for fast charging while limiting battery degradation mechanisms which shorten battery lifetime. Challenges in this optimization are the high dimensionality of the space of possible charging protocols, significant variability between batteries, and limited quantitative information on battery degradation mechanisms. Current approaches to addressing these challenges are model-based optimization and grid search. Optimization based on electrochemical models is limited by uncertainty in the underlying battery degradation mechanisms and grid search methods are expensive in terms of time, testing equipment, and cells. This article proposes a fast-charging Bayesian optimization strategy that explicitly includes constraints that limit degradation. The proposed BO-based charging approaches are sample-efficient and do not require first-principles models. Three different types of acquisition function (i.e., expected improvement, probability of improvement, and lower confidence bound) are evaluated. Their efficacies are compared for exploring and exploiting the parameter space of charging protocols for minimizing the charging time for lithium-ion batteries described by porous electrode theory. The probability-of-improvement acquisition function has lower mean and best minimum charging times than the lower-confidence-bound and expected-improvement acquisition functions. We quantify the decrease in the minimum charging time and increase in its uncertainty with increasing number of current steps used in charging protocols. Understanding ways to increase the convergence rate of Bayesian optimization, and how the convergence scales with the number of degrees of freedom in the optimization, serves as a baseline for extensions of the optimization to include battery design parameters. READ MORE
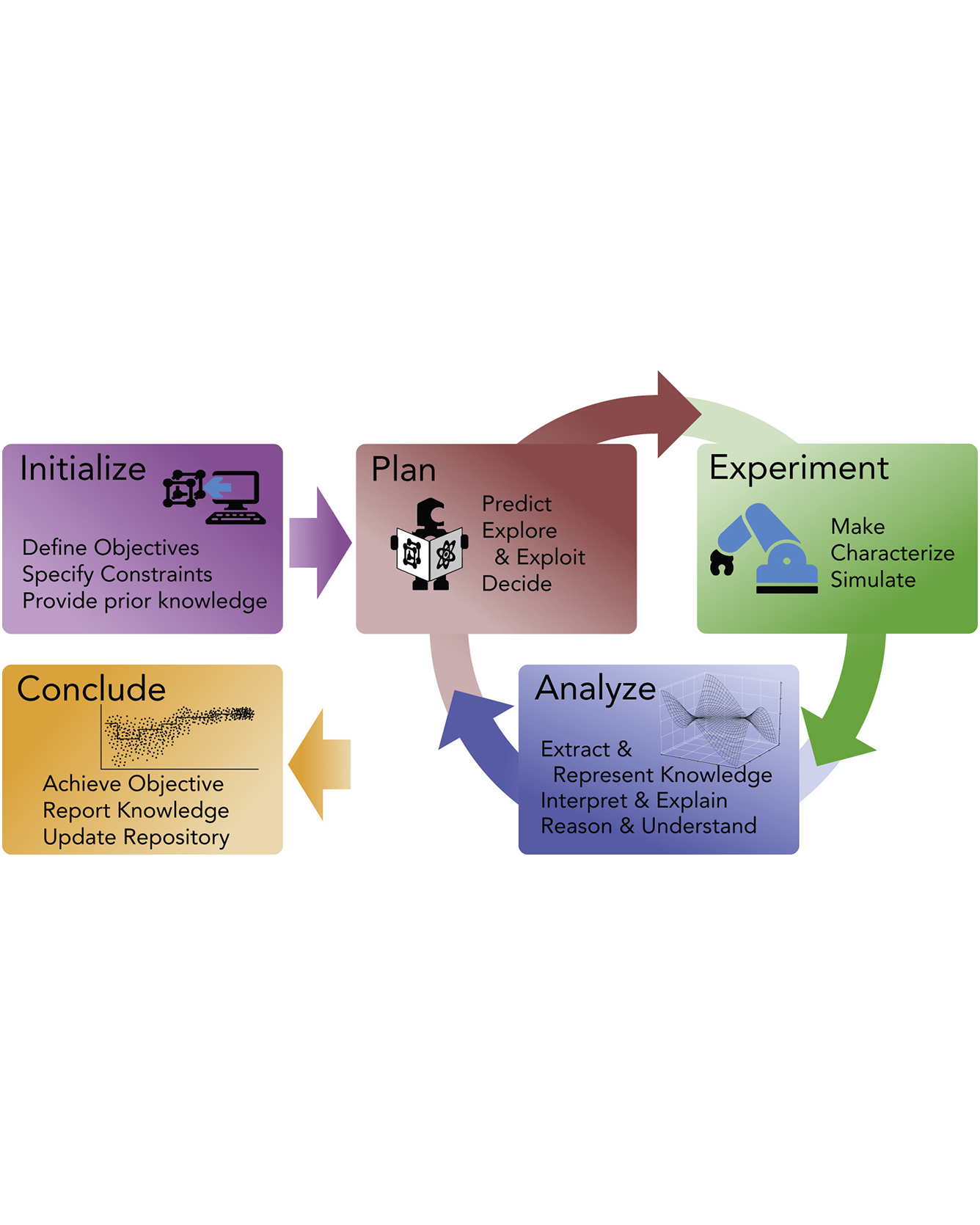
The modus operandi in materials research and development is combining existing data with an understanding of the underlying physics to create and test new hypotheses via experiments or simulations. This process is traditionally driven by subject expertise and the creativity of individual researchers, who “close the loop” by updating their hypotheses and models in light of new data or knowledge acquired from the community. Since the early 2000s, there has been notable progress in the automation of each step of the scientific process. With recent advances in using machine learning for hypothesis generation and artificial intelligence for decision-making, the opportunity to automate the entire closed-loop process has emerged as an exciting research frontier. The future of fully autonomous research systems for materials science no longer feels far-fetched. Autonomous systems are poised to make the search for new materials, properties, or parameters more efficient under budget and time constraints, and in effect accelerate materials innovation. This paper provides a brief overview of closed-loop research systems of today, and our related work at the Toyota Research Institute applied across different materials challenges and identifies both limitations and future opportunities. READ MORE
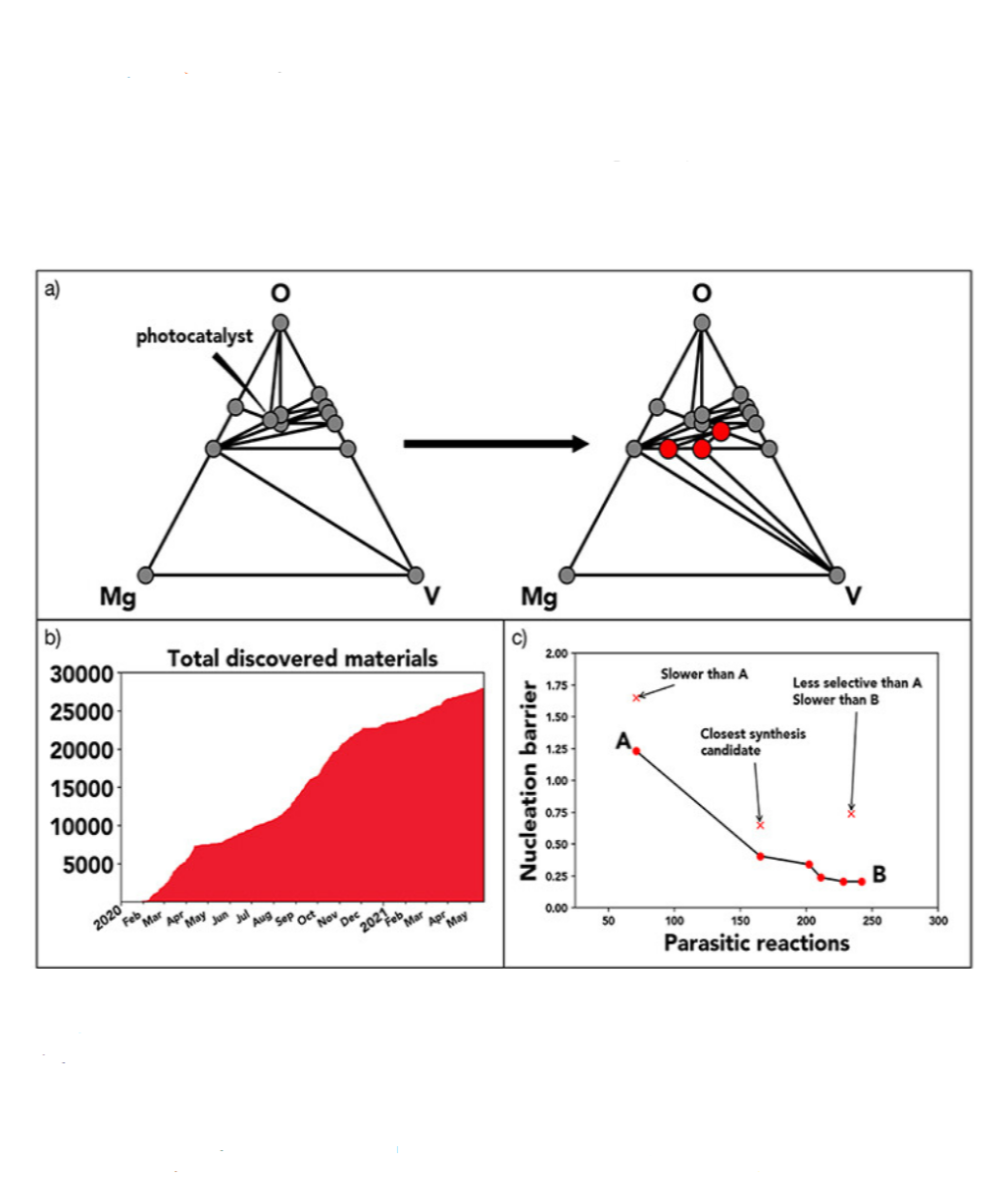
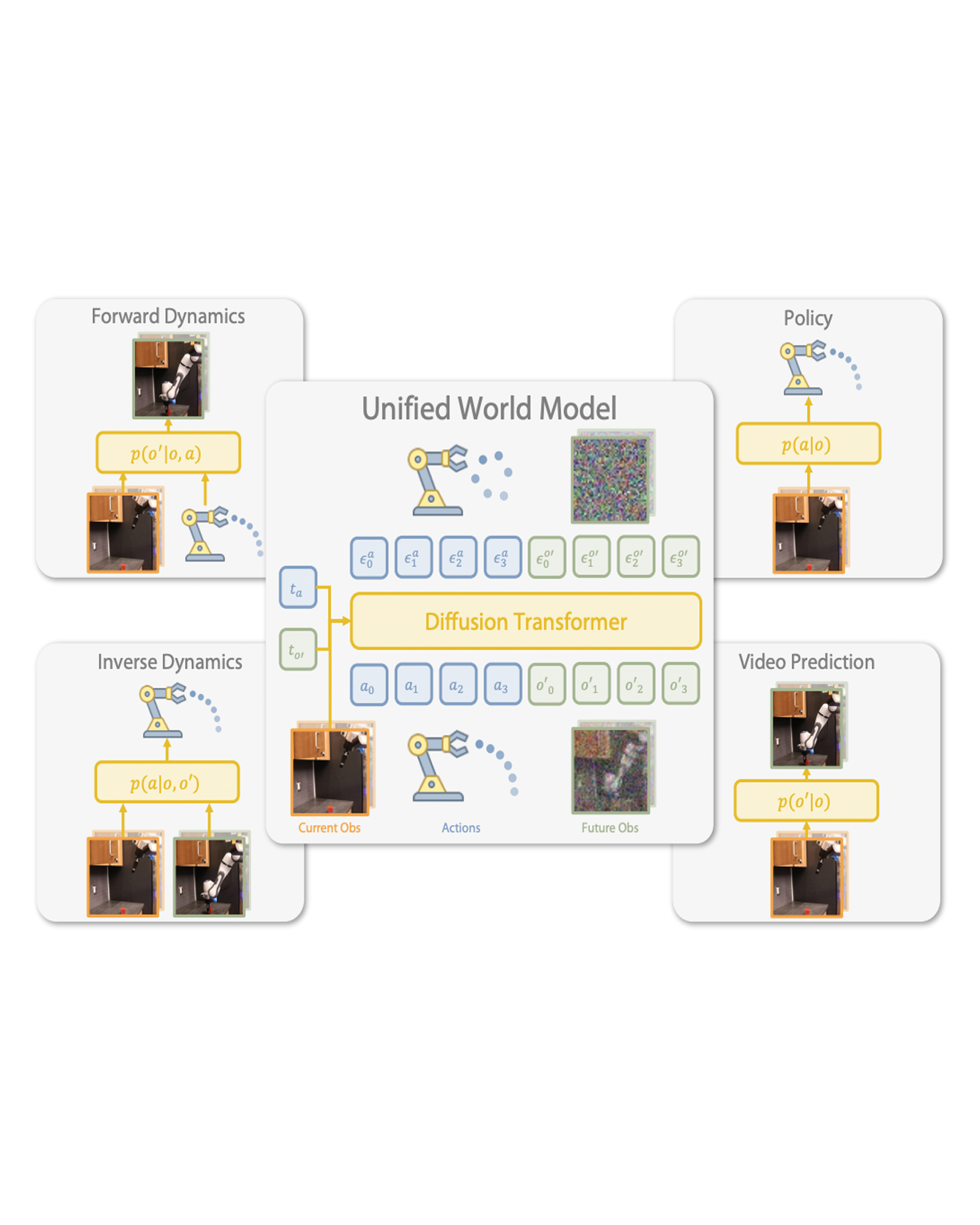
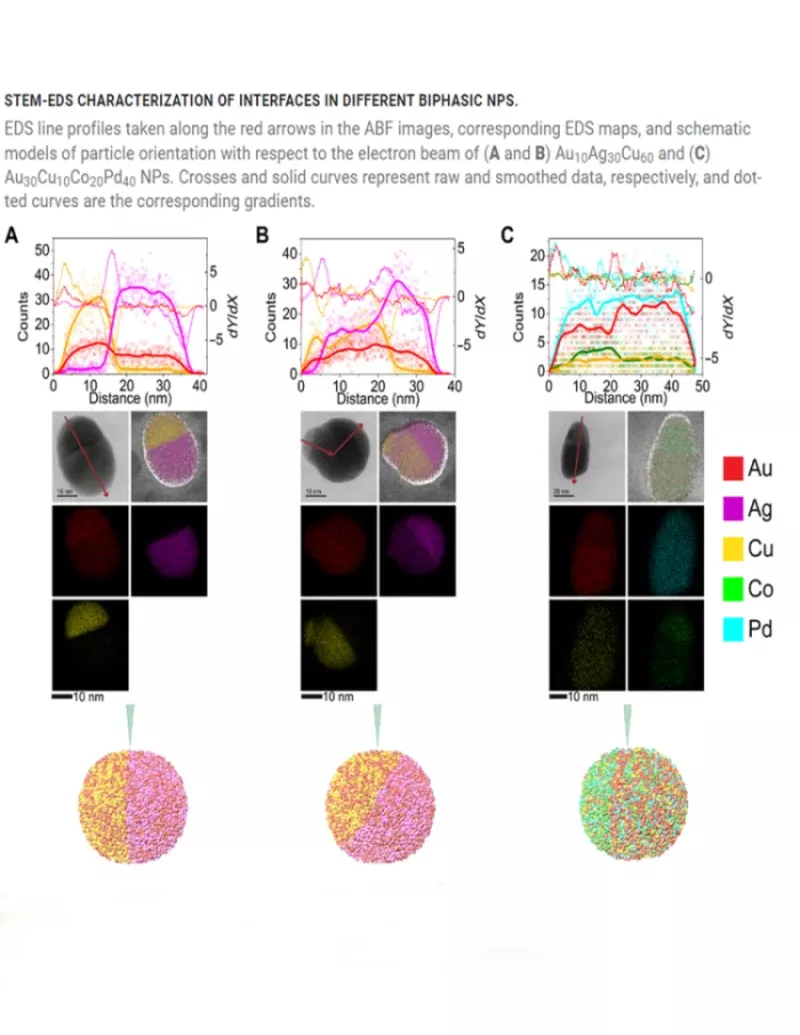
In materials discovery efforts, synthetic capabilities far outpace the ability to extract meaningful data from them. To bridge this gap, machine learning methods are necessary to reduce the search space for identifying desired materials. Here, we present a machine learning–driven, closed-loop experimental process to guide the synthesis of polyelemental nanomaterials with targeted structural properties. By leveraging data from an eight-dimensional chemical space (Au-Ag-Cu-Co-Ni-Pd-Sn-Pt) as inputs, a Bayesian optimization algorithm is used to suggest previously unidentified nanoparticle compositions that target specific interfacial motifs for synthesis, results of which are iteratively shared back with the algorithm. This feedback loop resulted in successful syntheses of 18 heterojunction nanomaterials that are too complex to discover by chemical intuition alone, including extremely chemically complex biphasic nanoparticles reported to date. Platforms like the one developed here are poised to transform materials discovery across a wide swath of applications and industries. READ MORE
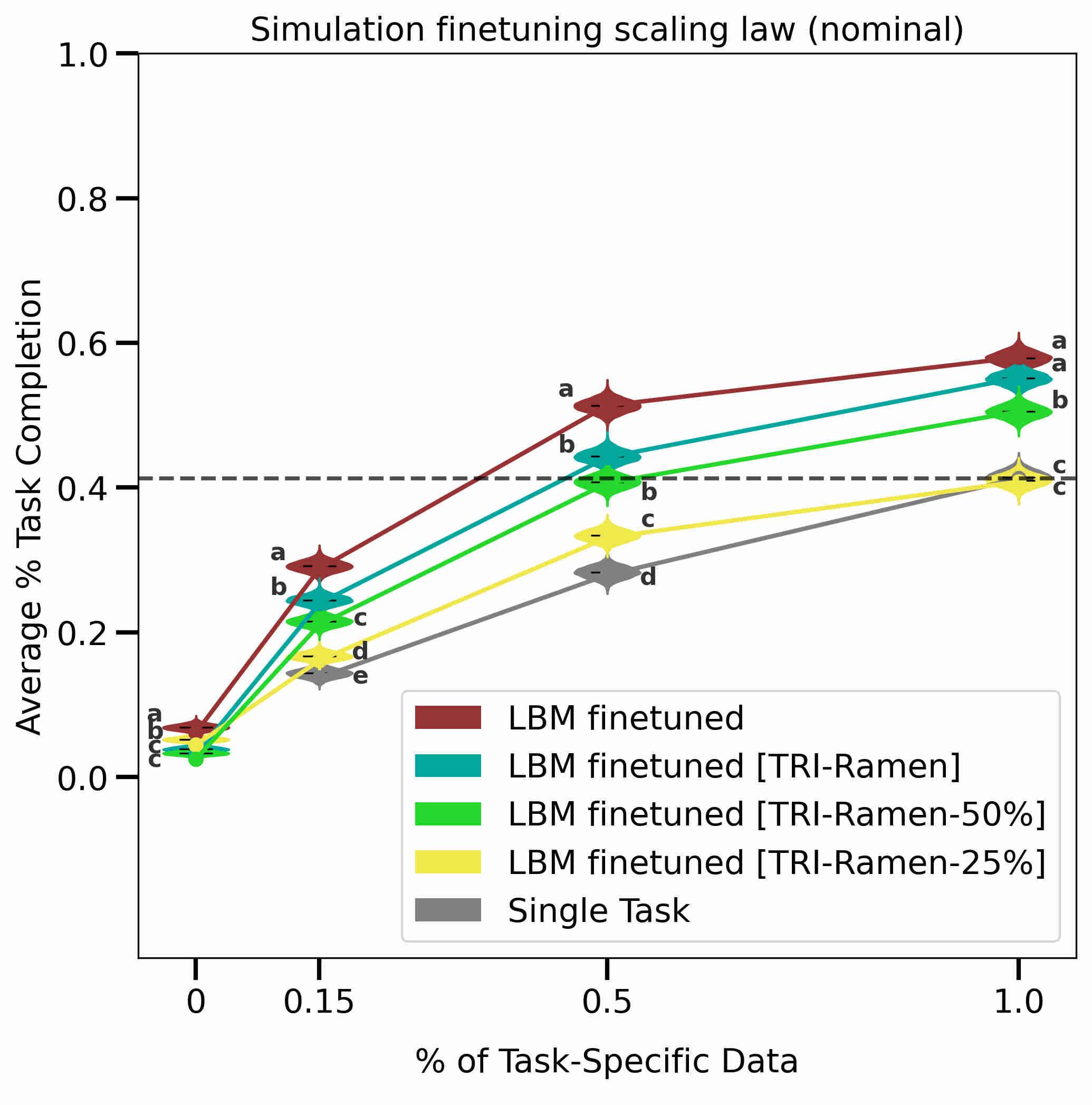
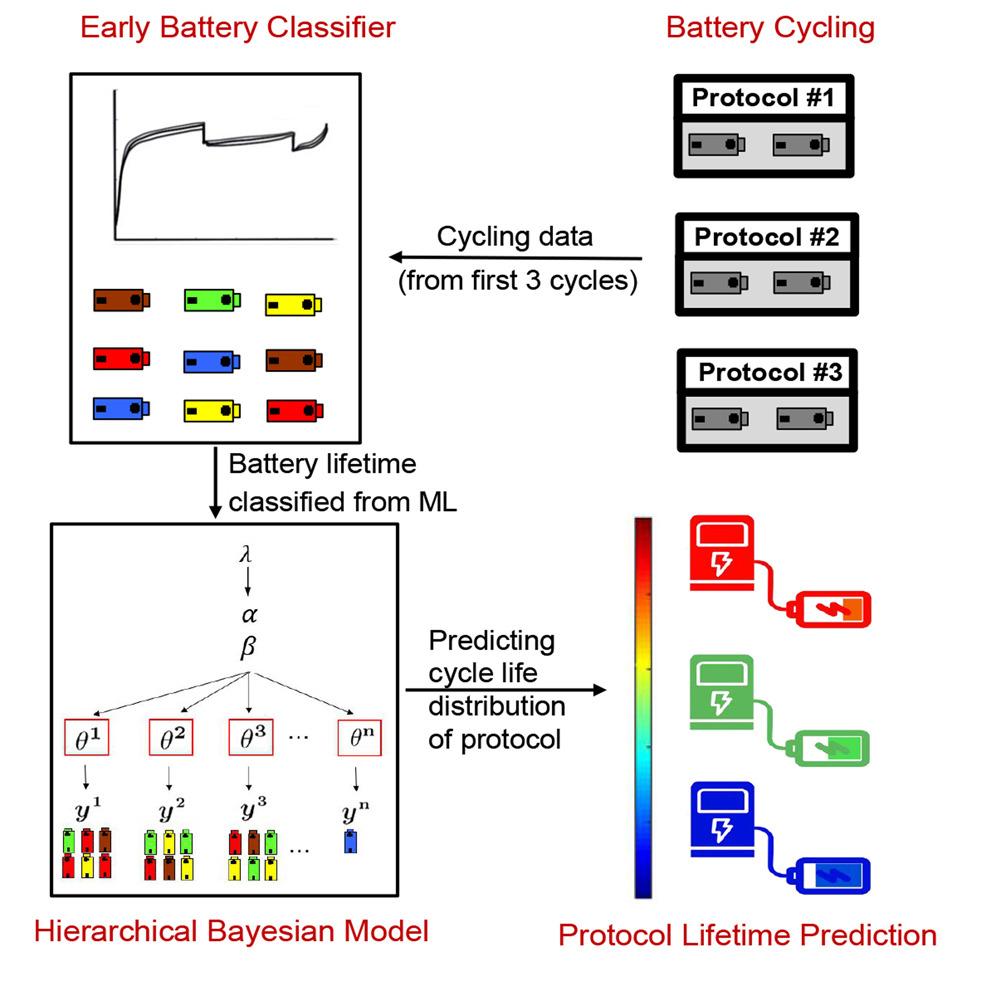
Advancing lithium-ion battery technology requires the optimization of cycling protocols. A new data-driven methodology is demonstrated for rapid, accurate prediction of the cycle life obtained by new cycling protocols using a single test lasting only 3 cycles, enabling rapid exploration of cycling protocol design spaces with orders of magnitude reduction in testing time. We achieve this by combining lifetime early prediction with a hierarchical Bayesian model (HBM) to rapidly predict performance distributions without the need for extensive repetitive testing. The methodology is applied to a comprehensive dataset of lithium-iron-phosphate/graphite comprising 29 different fast-charging protocols. HBM alone provides high protocol-lifetime prediction performance, with 6.5% of overall test average percent error, after cycling only one battery to failure. By combining HBM with a battery lifetime prediction model, we achieve a test error of 8.8% using a single 3-cycle test. In addition, the generalizability of the HBM approach is demonstrated for lithium-manganese-cobalt-oxide/graphite cells. READ MORE
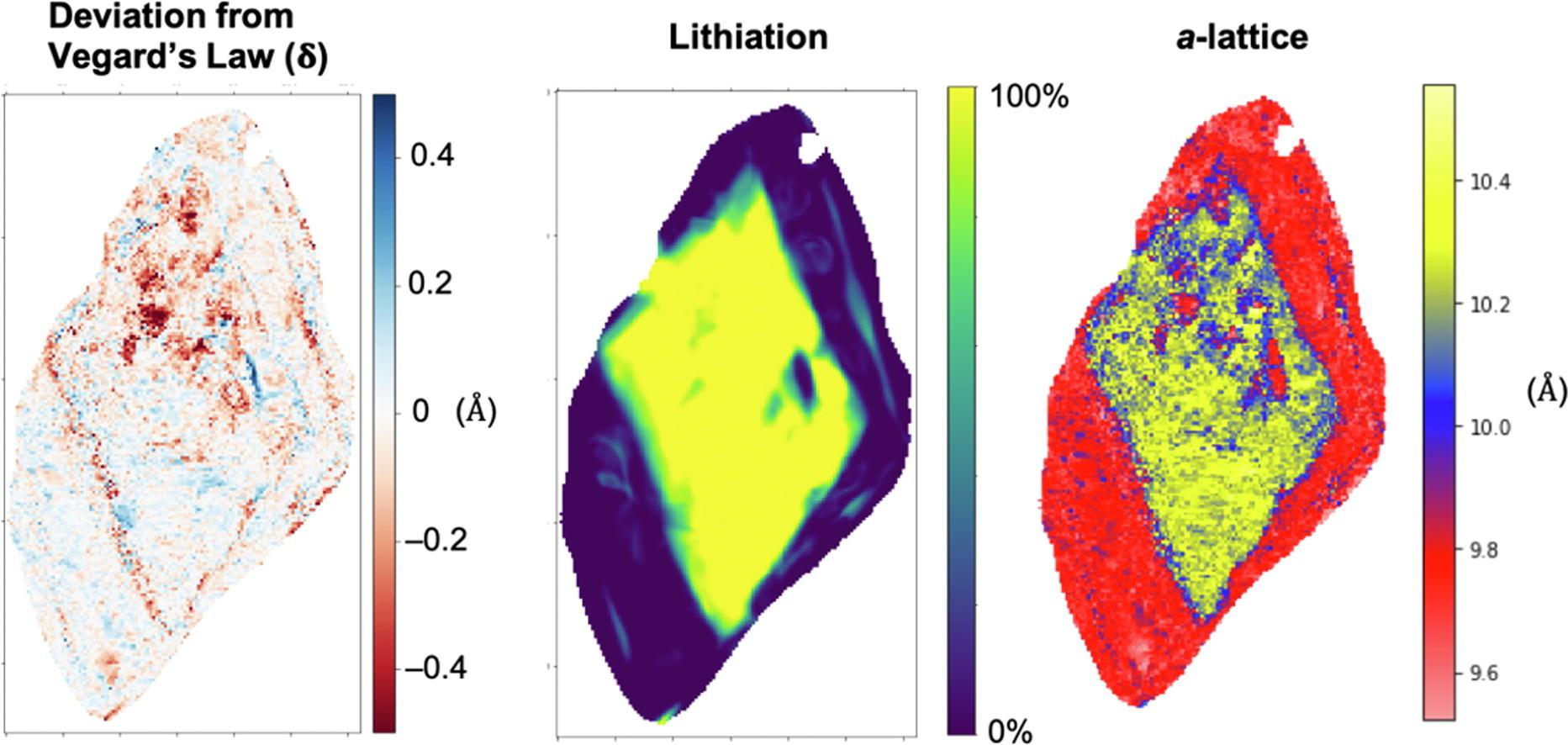
Lithium iron phosphate (LixFePO4), a cathode material used in rechargeable Li-ion batteries, phase separates upon de/lithiation under equilibrium. The interfacial structure and chemistry within these cathode materials affects Li-ion transport, and therefore battery performance. Correlative imaging of LixFePO4 was performed using four-dimensional scanning transmission electron microscopy (4D-STEM), scanning transmission X-ray microscopy (STXM), and X-ray ptychography in order to analyze the local structure and chemistry of the same particle set. Over 50,000 diffraction patterns from 10 particles provided measurements of both structure and chemistry at a nanoscale spatial resolution (16.6–49.5 nm) over wide (several micron) fields-of-view with statistical robustness. LixFePO4 particles at varying stages of delithiation were measured to examine the evolution of structure and chemistry as a function of delithiation. In lithiated and delithiated particles, local variations were observed in the degree of lithiation even while local lattice structures remained comparatively constant, and calculation of linear coefficients of chemical expansion suggest pinning of the lattice structures in these populations. Partially delithiated particles displayed broadly core–shell-like structures, however, with highly variable behavior both locally and per individual particle that exhibited distinctive intermediate regions at the interface between phases, and pockets within the lithiated core that correspond to FePO4 in structure and chemistry. The results provide insight into the LixFePO4 system, subtleties in the scope and applicability of Vegard’s law (linear lattice parameter-composition behavior) under local versus global measurements, and demonstrate a powerful new combination of experimental and analytical modalities for bridging the crucial gap between local and statistical characterization. READ MORE
Solutions to many of the world's problems depend upon materials research and development. However, advanced materials can take decades to discover and decades more to fully deploy. Humans and robots have begun to partner to advance science and technology orders of magnitude faster than humans do today through the development and exploitation of closed-loop, autonomous experimentation systems. This review discusses the specific challenges and opportunities related to materials discovery and development that will emerge from this new paradigm. Our perspective incorporates input from stakeholders in academia, industry, government laboratories, and funding agencies. We outline the current status, barriers, and needed investments, culminating with a vision for the path forward. We intend the article to spark interest in this emerging research area and to motivate potential practitioners by illustrating early successes. We also aspire to encourage a creative reimagining of the next generation of materials science infrastructure. To this end, we frame future investments in materials science and technology, hardware and software infrastructure, artificial intelligence and autonomy methods, and critical workforce development for autonomous research. READ MORE