Featured Publications

All Publications
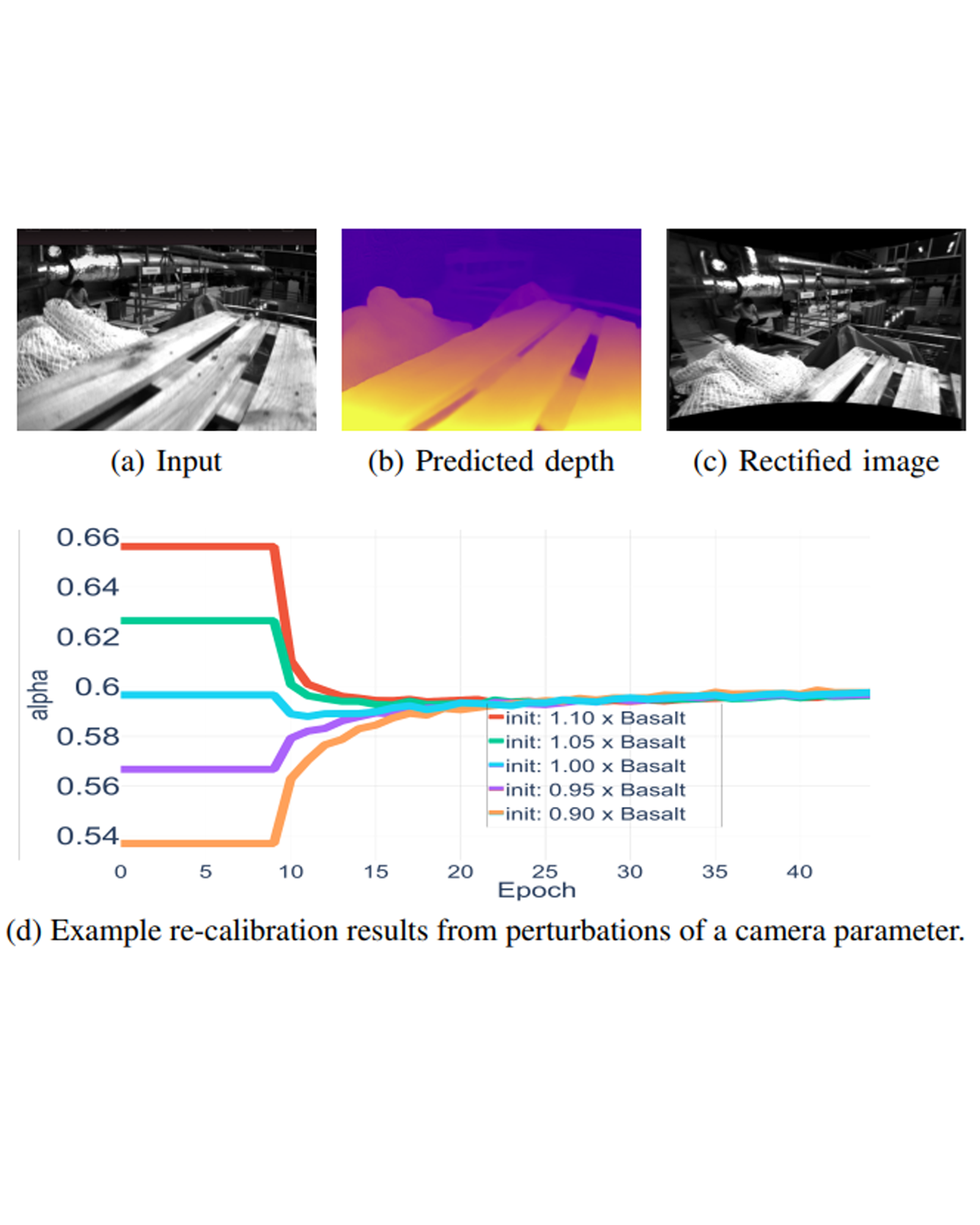
Camera calibration is integral to robotics and computer vision algorithms that seek to infer geometric properties of the scene from visual input streams. In practice, calibration is a laborious procedure requiring specialized data collection and careful tuning. This process must be repeated whenever the parameters of the camera change, which can be a frequent occurrence for mobile robots and autonomous vehicles. In contrast, self-supervised depth and ego-motion estimation approaches can bypass explicit calibration by inferring per-frame projection models that optimize a view synthesis objective. In this paper, we extend this approach to explicitly calibrate a wide range of cameras from raw videos in the wild. We propose a learning algorithm to regress per-sequence calibration parameters using an efficient family of general camera models. Our procedure achieves self-calibration results with sub-pixel reprojection error, outperforming other learning-based methods. We validate our approach on a wide variety of camera geometries, including perspective, fisheye, and catadioptric. Finally, we show that our approach leads to improvements in the downstream task of depth estimation, achieving state-of-the-art results on the EuRoC dataset with greater computational efficiency than contemporary methods. READ MORE
Chapter 3
The materials discovery process naturally presents a slew of questions about the character of a material. These range from simply trying to learn basic facts about the atomic structure of the compound [1–3] all the way to producing a time-resolved profile of a functional process in operando from start to finish [4]. Spectros-copy is the process of measuring a materials’ response to external electromagnetic stimulus to deduce the properties of interest. Spectroscopy can help to solve problems in experimental design and decision-making (probing response of certain energy domains, measuring particular properties), inference (moving from raw data to the property of interest), and analysis (rationalizing and interpreting the data). These are all classes of problems which artificial intelligence (AI) is well-posed to address.As such, the interface between practitioners in both spectroscopy and AI has sparked a great deal of excitement and research activity. Because characterization is a crucial process of the materials discovery pipeline, insightful pairings of algorithms and experimental protocols can lead to gains in both experimental efficiency and accuracy.These gains may significantly compress the timescales of experimental procedures and help experimentalists learn more in less time or answer questions which were previously inaccessible with a given experimental apparatus. READ MORE

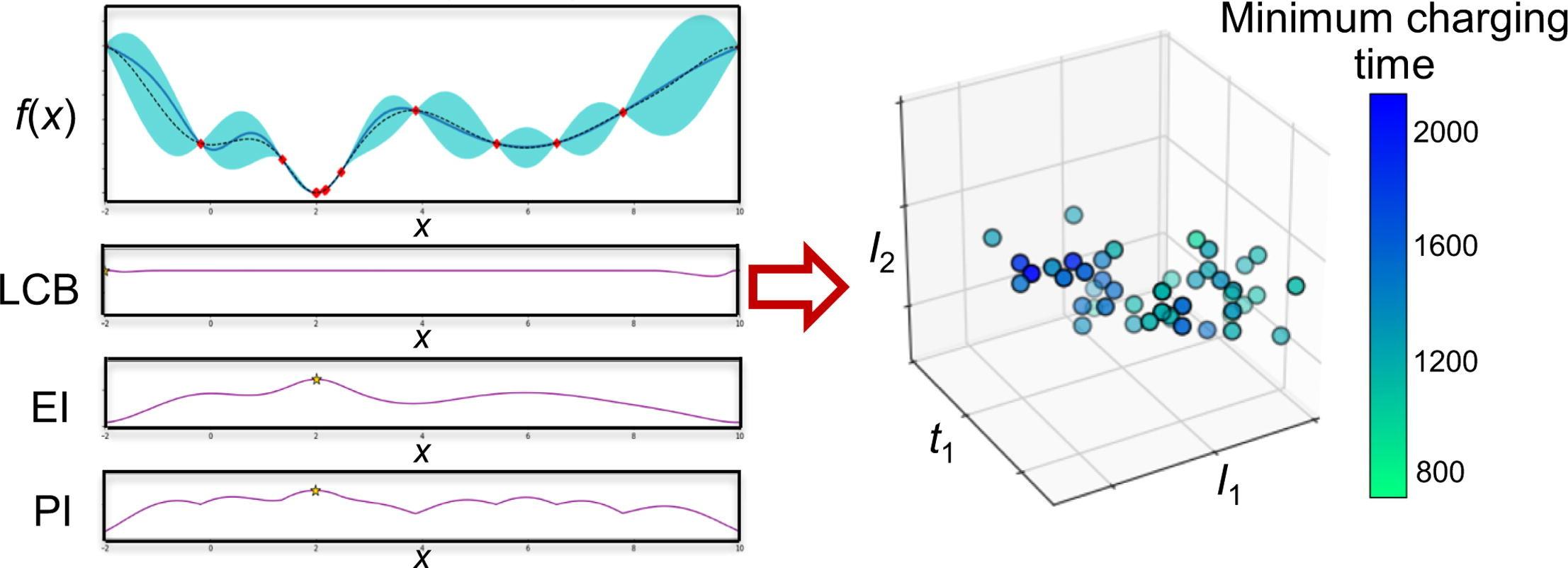
Lithium-ion batteries are one of the most commonly used energy storage device for electric vehicles. As battery chemistries continue to advance, an important question concerns how to efficiently determine charging protocols that best balance the desire for fast charging while limiting battery degradation mechanisms which shorten battery lifetime. Challenges in this optimization are the high dimensionality of the space of possible charging protocols, significant variability between batteries, and limited quantitative information on battery degradation mechanisms. Current approaches to addressing these challenges are model-based optimization and grid search. Optimization based on electrochemical models is limited by uncertainty in the underlying battery degradation mechanisms and grid search methods are expensive in terms of time, testing equipment, and cells. This article proposes a fast-charging Bayesian optimization strategy that explicitly includes constraints that limit degradation. The proposed BO-based charging approaches are sample-efficient and do not require first-principles models. Three different types of acquisition function (i.e., expected improvement, probability of improvement, and lower confidence bound) are evaluated. Their efficacies are compared for exploring and exploiting the parameter space of charging protocols for minimizing the charging time for lithium-ion batteries described by porous electrode theory. The probability-of-improvement acquisition function has lower mean and best minimum charging times than the lower-confidence-bound and expected-improvement acquisition functions. We quantify the decrease in the minimum charging time and increase in its uncertainty with increasing number of current steps used in charging protocols. Understanding ways to increase the convergence rate of Bayesian optimization, and how the convergence scales with the number of degrees of freedom in the optimization, serves as a baseline for extensions of the optimization to include battery design parameters. READ MORE
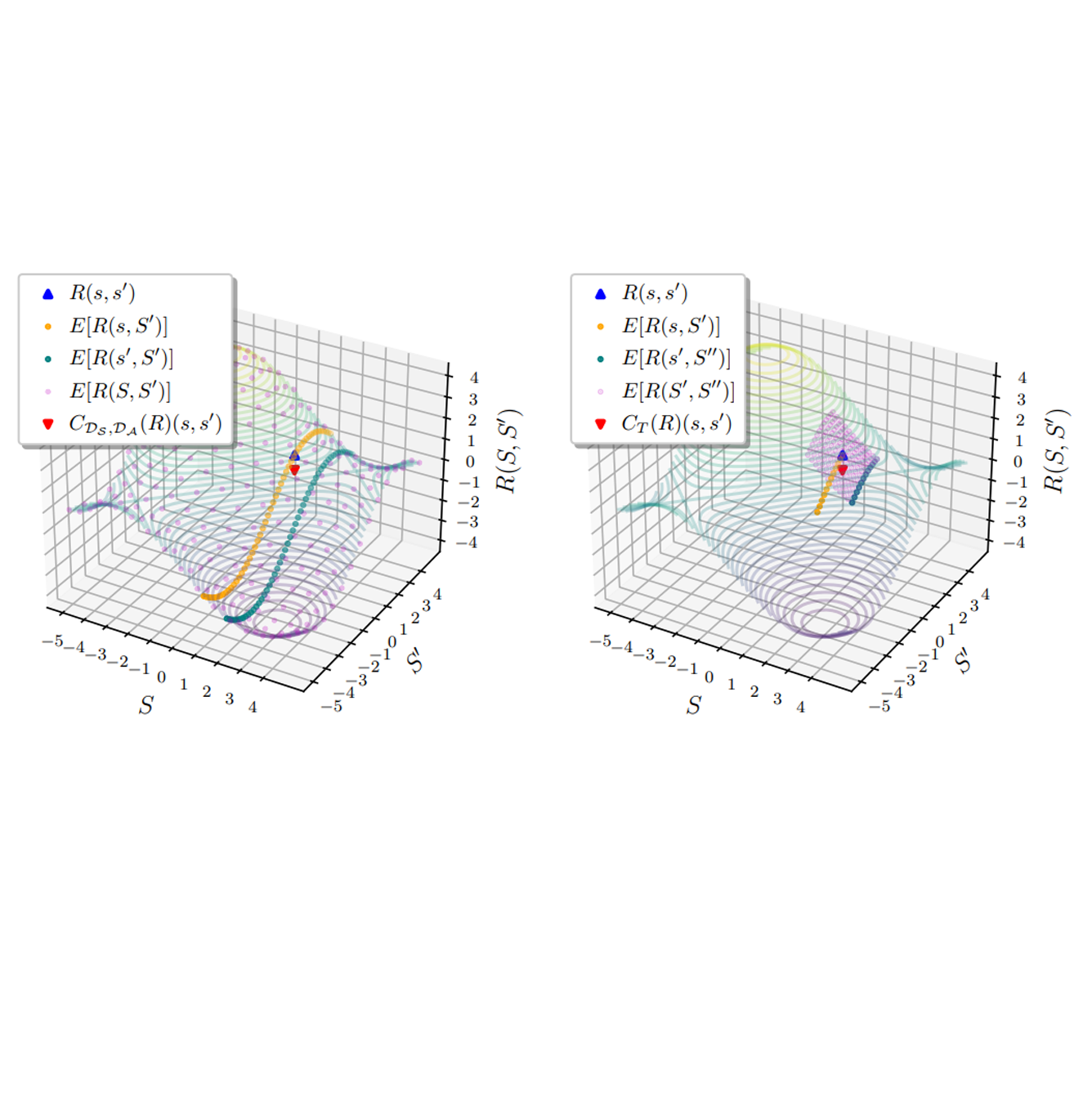
The ability to learn reward functions plays an important role in enabling the deployment of intelligent agents in the real world. However, comparing reward functions, for example as a means of evaluating reward learning methods, presents a challenge. Reward functions are typically compared by considering the behavior of optimized policies, but this approach conflates deficiencies in the reward function with those of the policy search algorithm used to optimize it. To address this challenge, Gleave et al. (2020) propose the Equivalent-Policy Invariant Comparison (EPIC) distance. EPIC avoids policy optimization, but in doing so requires computing reward values at transitions that may be impossible under the system dynamics. This is problematic for learned reward functions because it entails evaluating them outside of their training distribution, resulting in inaccurate reward values that we show can render EPIC ineffective at comparing rewards. To address this problem, we propose the Dynamics-Aware Reward Distance (DARD), a new reward pseudometric. DARD uses an approximate transition model of the environment to transform reward functions into a form that allows for comparisons that are invariant to reward shaping while only evaluating reward functions on transitions close to their training distribution. Experiments in simulated physical domains demonstrate that DARD enables reliable reward comparisons without policy optimization and is significantly more predictive than baseline methods of downstream policy performance when dealing with learned reward functions. READ MORE
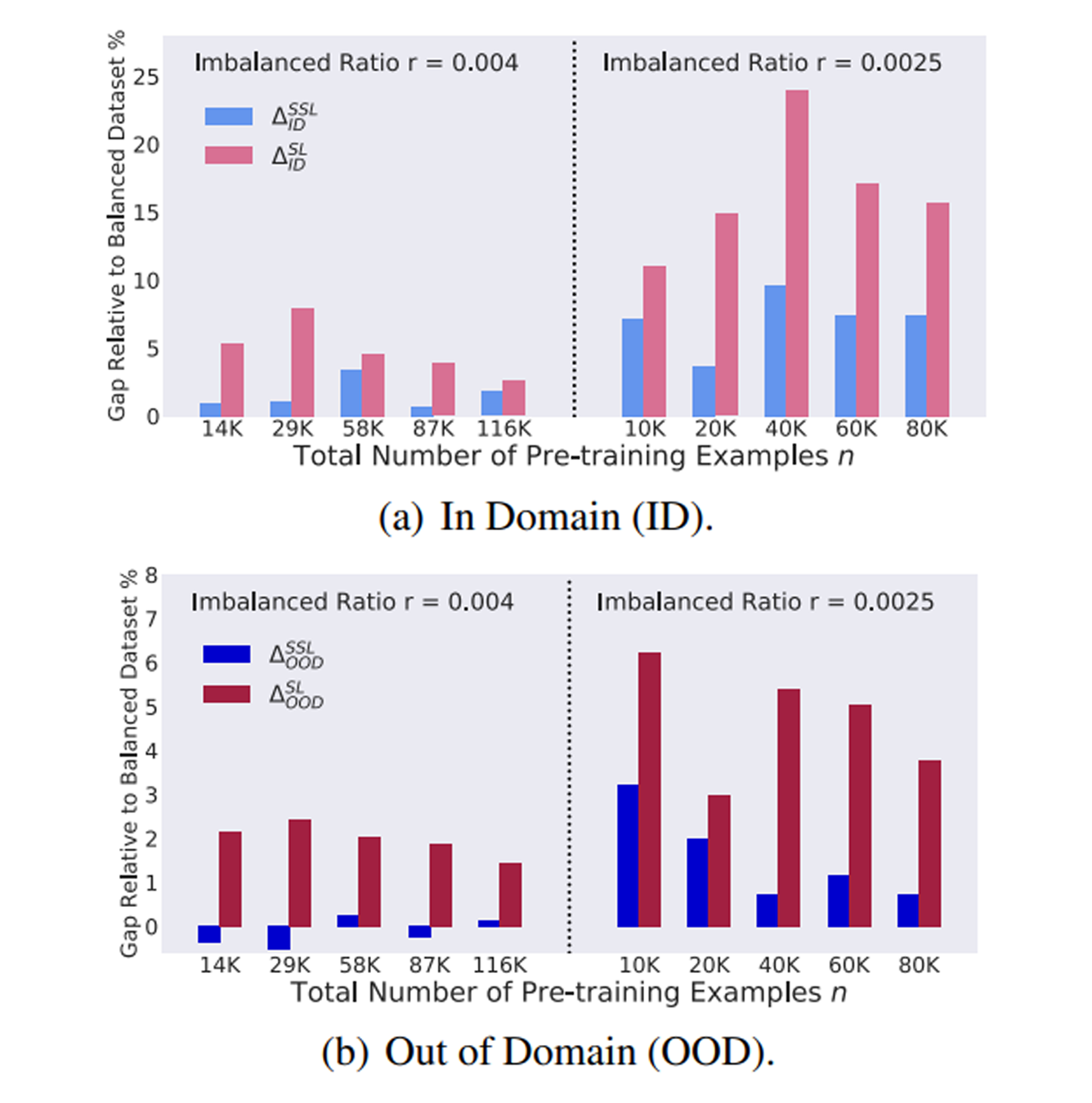
Self-supervised learning (SSL) is a scalable way to learn general visual representations since it learns without labels. However, large-scale unlabeled datasets in the wild often have long-tailed label distributions, where we know little about the behavior of SSL. In this work, we systematically investigate self-supervised learning under dataset imbalance. First, we find out via extensive experiments that off-the-shelf self-supervised representations are already more robust to class imbalance than supervised representations. The performance gap between balanced and imbalanced pre-training with SSL is significantly smaller than the gap with supervised learning, across sample sizes, for both in-domain and, especially, out-of-domain evaluation. Second, towards understanding the robustness of SSL, we hypothesize that SSL learns richer features from frequent data: it may learn label-irrelevant-but-transferable features that help classify the rare classes and downstream tasks. In contrast, supervised learning has no incentive to learn features irrelevant to the labels from frequent examples. We validate this hypothesis with semi-synthetic experiments and theoretical analyses on a simplified setting. Third, inspired by the theoretical insights, we devise a re-weighted regularization technique that consistently improves the SSL representation quality on imbalanced datasets with several evaluation criteria, closing the small gap between balanced and imbalanced datasets with the same number of examples. READ MORE
The modus operandi in materials research and development is combining existing data with an understanding of the underlying physics to create and test new hypotheses via experiments or simulations. This process is traditionally driven by subject expertise and the creativity of individual researchers, who “close the loop” by updating their hypotheses and models in light of new data or knowledge acquired from the community. Since the early 2000s, there has been notable progress in the automation of each step of the scientific process. With recent advances in using machine learning for hypothesis generation and artificial intelligence for decision-making, the opportunity to automate the entire closed-loop process has emerged as an exciting research frontier. The future of fully autonomous research systems for materials science no longer feels far-fetched. Autonomous systems are poised to make the search for new materials, properties, or parameters more efficient under budget and time constraints, and in effect accelerate materials innovation. This paper provides a brief overview of closed-loop research systems of today, and our related work at the Toyota Research Institute applied across different materials challenges and identifies both limitations and future opportunities. READ MORE
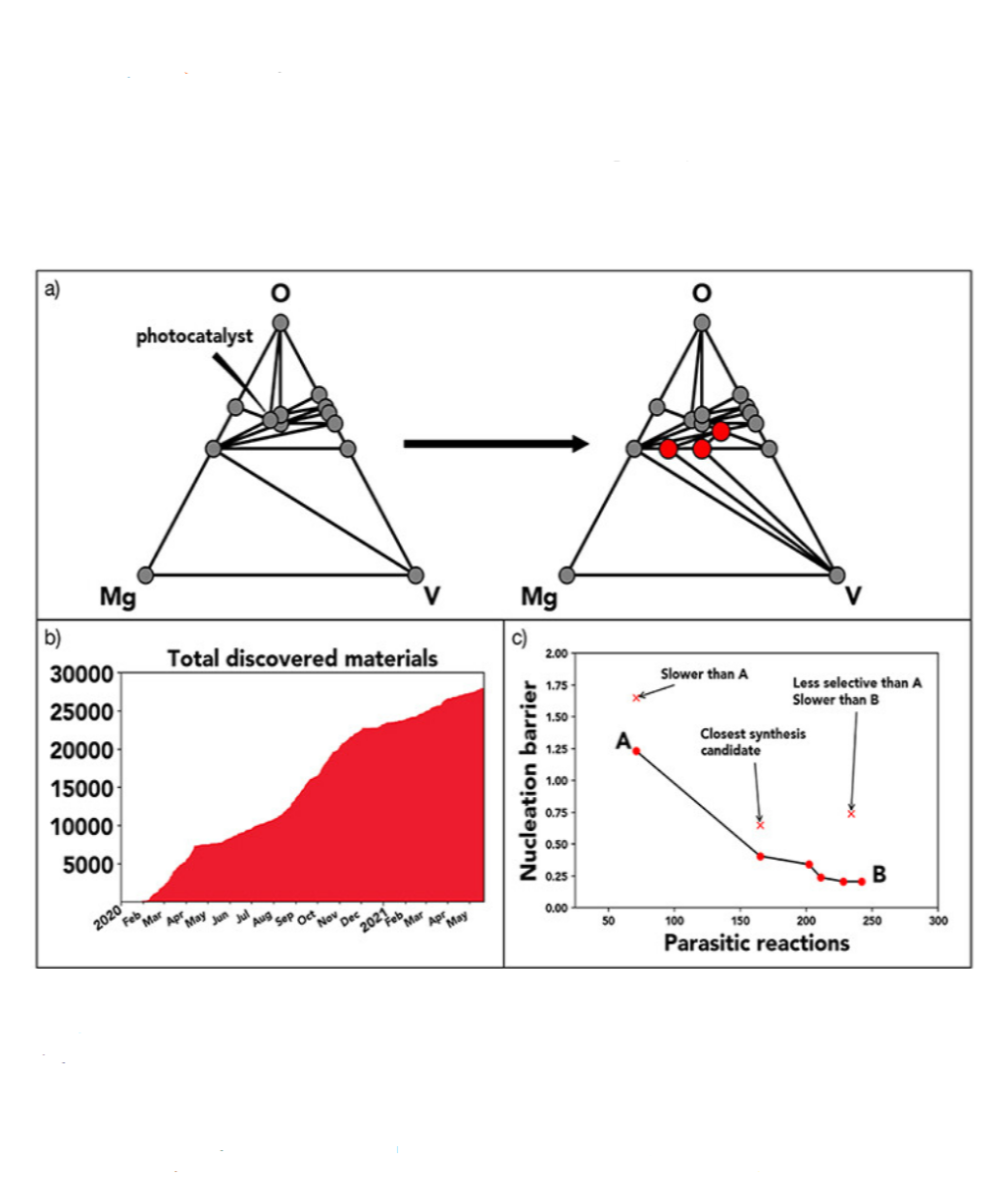
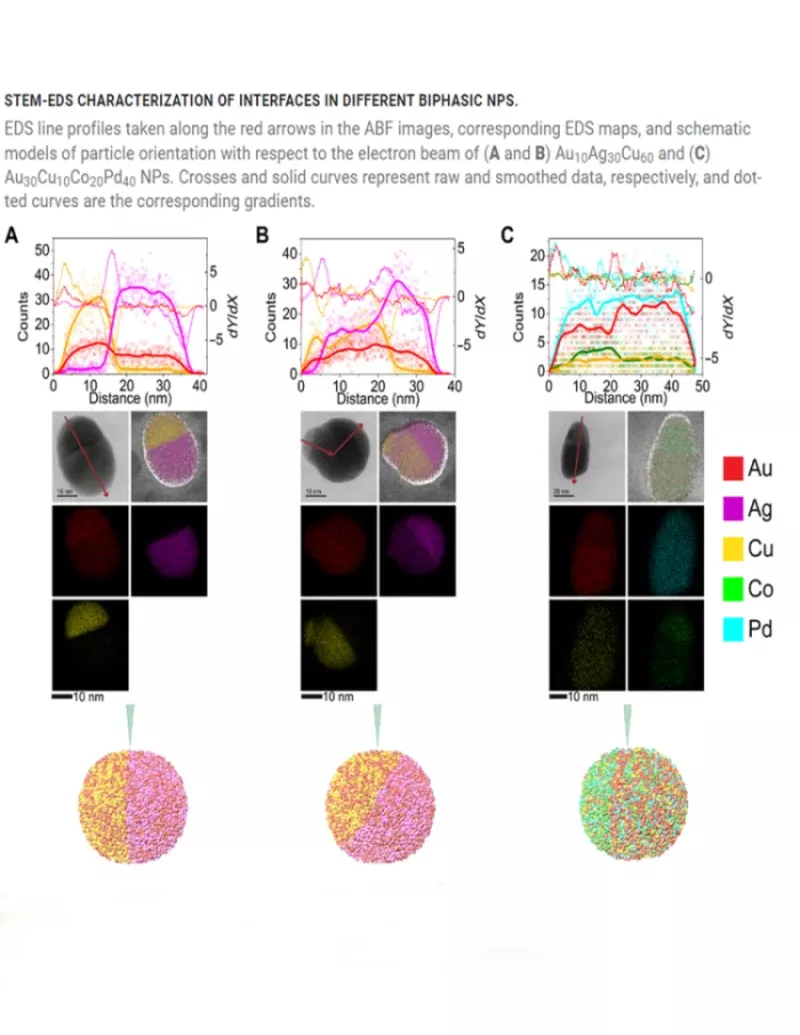
In materials discovery efforts, synthetic capabilities far outpace the ability to extract meaningful data from them. To bridge this gap, machine learning methods are necessary to reduce the search space for identifying desired materials. Here, we present a machine learning–driven, closed-loop experimental process to guide the synthesis of polyelemental nanomaterials with targeted structural properties. By leveraging data from an eight-dimensional chemical space (Au-Ag-Cu-Co-Ni-Pd-Sn-Pt) as inputs, a Bayesian optimization algorithm is used to suggest previously unidentified nanoparticle compositions that target specific interfacial motifs for synthesis, results of which are iteratively shared back with the algorithm. This feedback loop resulted in successful syntheses of 18 heterojunction nanomaterials that are too complex to discover by chemical intuition alone, including extremely chemically complex biphasic nanoparticles reported to date. Platforms like the one developed here are poised to transform materials discovery across a wide swath of applications and industries. READ MORE
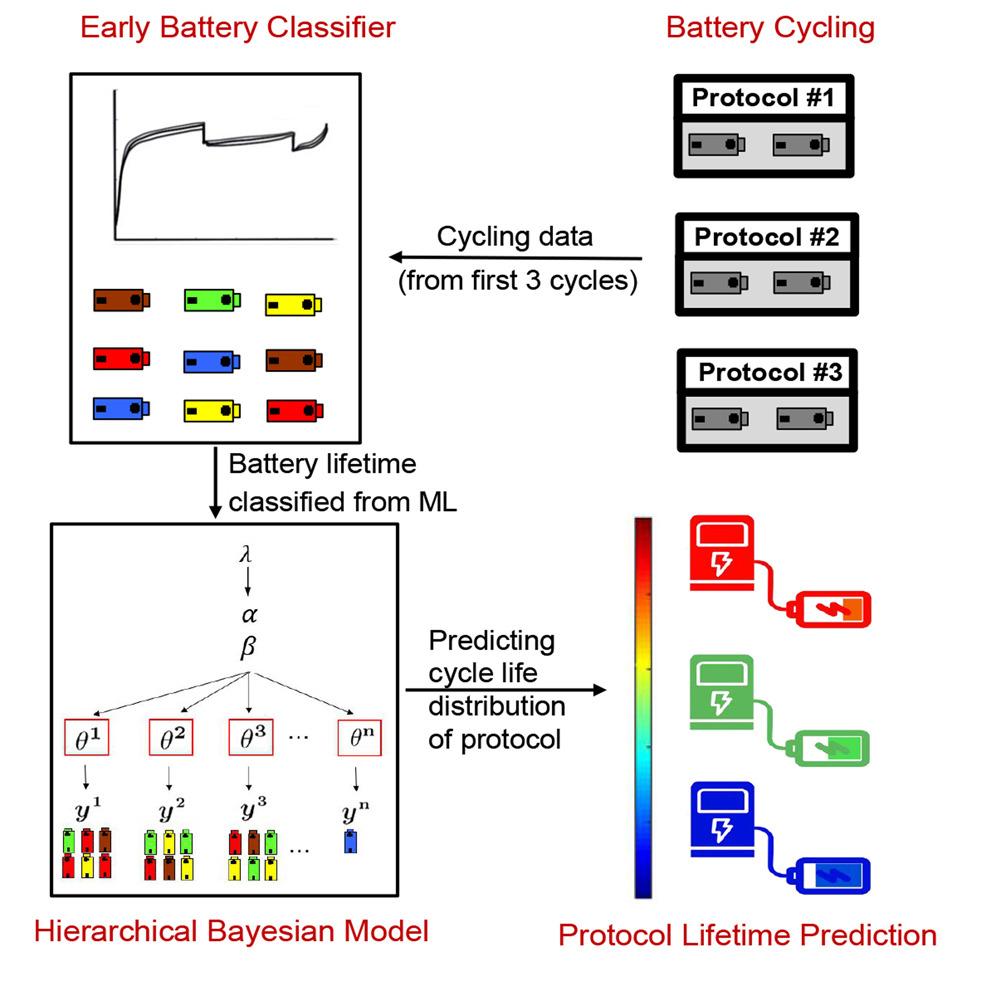
Advancing lithium-ion battery technology requires the optimization of cycling protocols. A new data-driven methodology is demonstrated for rapid, accurate prediction of the cycle life obtained by new cycling protocols using a single test lasting only 3 cycles, enabling rapid exploration of cycling protocol design spaces with orders of magnitude reduction in testing time. We achieve this by combining lifetime early prediction with a hierarchical Bayesian model (HBM) to rapidly predict performance distributions without the need for extensive repetitive testing. The methodology is applied to a comprehensive dataset of lithium-iron-phosphate/graphite comprising 29 different fast-charging protocols. HBM alone provides high protocol-lifetime prediction performance, with 6.5% of overall test average percent error, after cycling only one battery to failure. By combining HBM with a battery lifetime prediction model, we achieve a test error of 8.8% using a single 3-cycle test. In addition, the generalizability of the HBM approach is demonstrated for lithium-manganese-cobalt-oxide/graphite cells. READ MORE
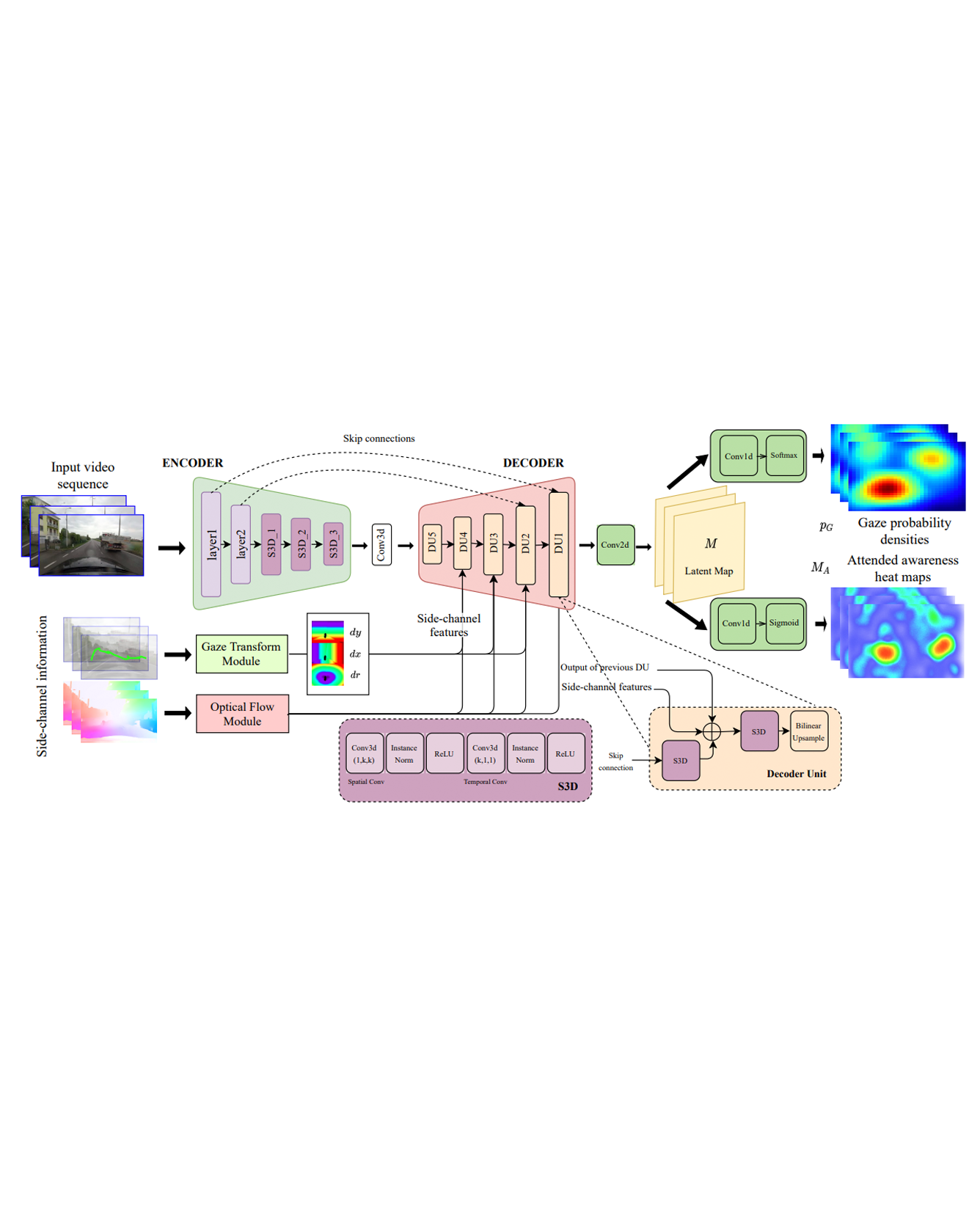
We propose a computational model to estimate a person’s attended awareness of their environment. We define “attended awareness” to be those parts of a potentially dynamic scene which a person has attended to in recent history and which they are still likely to be physically aware of. Our model takes as input scene information in the form of a video and noisy gaze estimates, and outputs visual saliency, a refined gaze estimate and an estimate of the person’s attended awareness. In order to test our model, we capture a new dataset with a high-precision gaze tracker including 24.5 hours of gaze sequences from 23 subjects attending to videos of driving scenes. The dataset also contains third-party annotations of the subjects’ attended awareness based on observations of their scan path. Our results show that our model is able to reasonably estimate attended awareness in a controlled setting, and in the future could potentially be extended to real egocentric driving data to help enable more effective ahead-of-time warnings in safety systems and thereby augment driver performance. We also demonstrate our model’s effectiveness on the tasks of saliency, gaze calibration and denoising, using both our dataset and an existing saliency dataset. We make our model and dataset available at https://github.com/ToyotaResearchInstitute/att-aware/. READ MORE
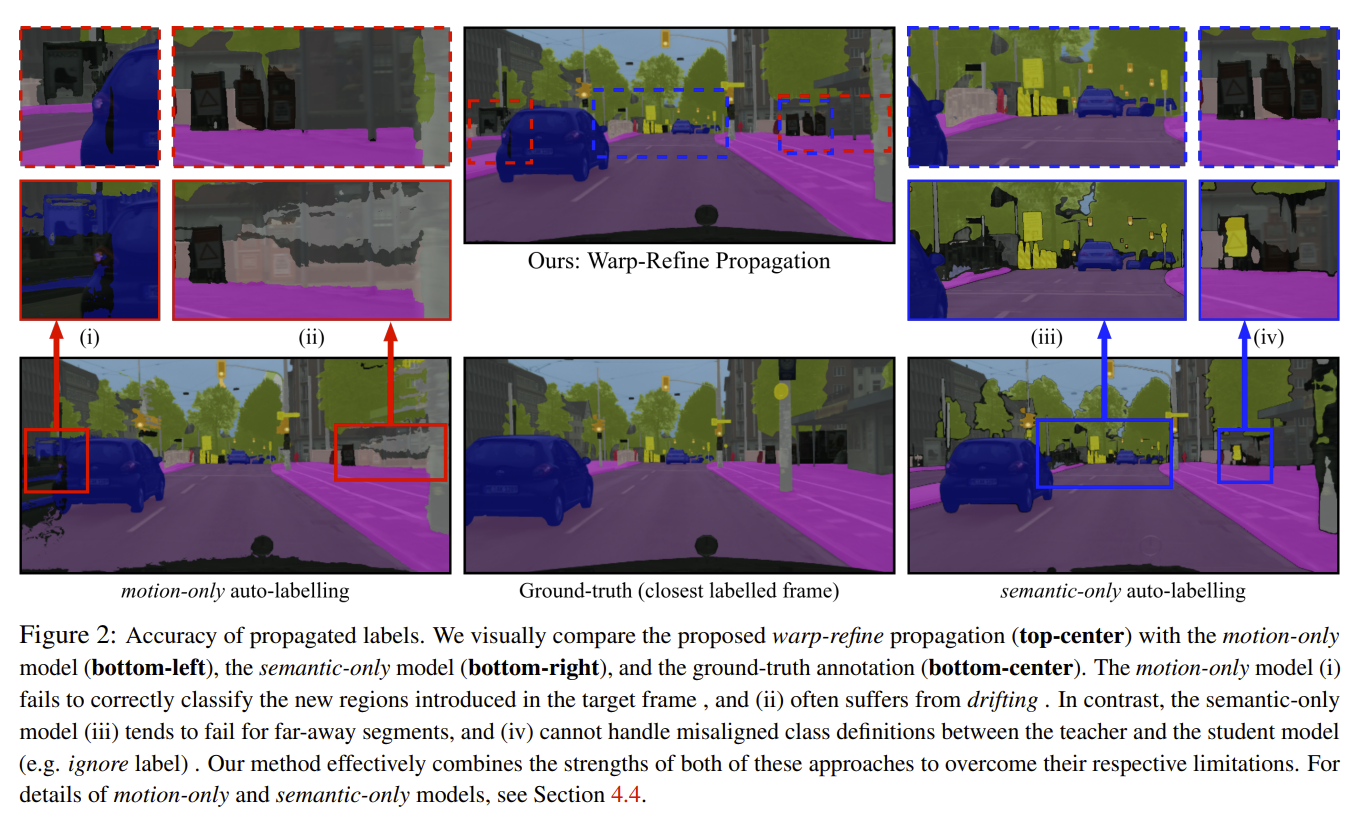
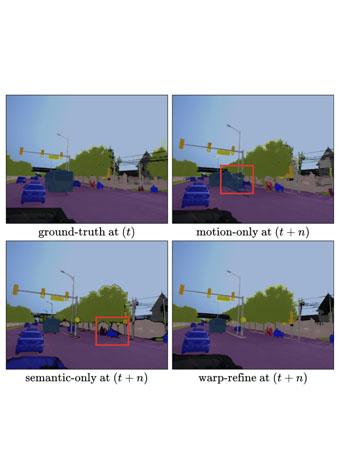
Deep learning models for semantic segmentation rely on expensive, large-scale, manually annotated datasets. Labelling is a tedious process that can take hours per image. Automatically annotating video sequences by propagating sparsely labeled frames through time is a more scalable alternative. In this work, we propose a novel label propagation method, termed Warp-Refine Propagation, that combines semantic cues with geometric cues to efficiently auto-label videos. Our method learns to refine geometrically-warped labels and infuse them with learned semantic priors in a semi-supervised setting by leveraging cycle consistency across time. We quantitatively show that our method improves label-propagation by a noteworthy margin of 13.1 mIoU on the ApolloScape dataset. Furthermore, by training with the auto-labelled frames, we achieve competitive results on three semantic-segmentation benchmarks, improving the state-of-the-art by a large margin of 1.8 and 3.61 mIoU on NYU-V2 and KITTI, while matching the current best results on Cityscapes. READ MORE