Featured Publications

All Publications
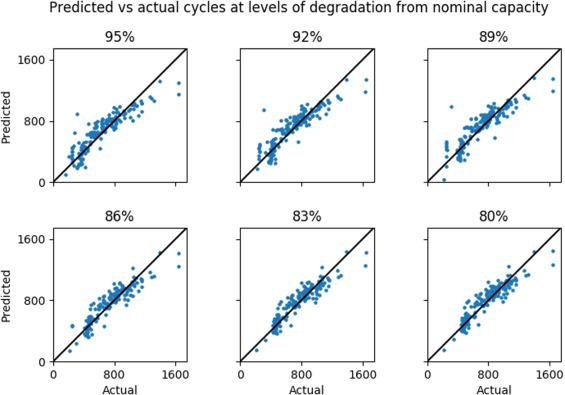
Battery evaluation and early prediction software package (BEEP) provides an open-source Python-based framework for the management and processing of high-throughput battery cycling data-streams. BEEPs features include file-system based organization of raw cycling data and metadata received from cell testing equipment, validation protocols that ensure the integrity of such data, parsing and structuring of data into Python-objects ready for analytics, featurization of structured cycling data to serve as input for machine-learning, and end-to-end examples that use processed data for anomaly detection and featurized data to train early-prediction models for cycle life. BEEP is developed in response to the software and expertise gap between cell-level battery testing and data-driven battery development. READ MORE
TRI Author: Joseph Montoya
All Authors: Anjli Patel, Jens Nørskov, Kristin Persson, Joseph Montoya
Pourbaix diagrams have been used extensively to evaluate stability regions of materials subject to varying potential and pH conditions in aqueous environments. However, both recent advances in high-throughput material exploration and increasing complexity of materials of interest for electrochemical applications pose challenges for performing Pourbaix analysis on multidimensional systems. Specifically, current Pourbaix construction algorithms incur significant computational costs for systems consisting of four or more elemental components. Herein, we propose an alternative Pourbaix construction method that filters all potential combinations of species in a system to only those present on a compositional convex hull. By including axes representing the quantities of H+ and e− required to form a given phase, one can ensure every stable phase mixture is included in the Pourbaix diagram and reduce the computational time required to construct the resultant Pourbaix diagram by several orders of magnitude. This new Pourbaix algorithm has been incorporated into the pymatgen code and the Materials Project website, and it extends the ability to evaluate the Pourbaix stability of complex multicomponent systems. Read More
Citation: Patel, Anjli M., Jens K. Nørskov, Kristin A. Persson, and Joseph H. Montoya. "Efficient Pourbaix diagrams of many-element compounds." Physical Chemistry Chemical Physics 21, no. 45 (2019): 25323-25327.
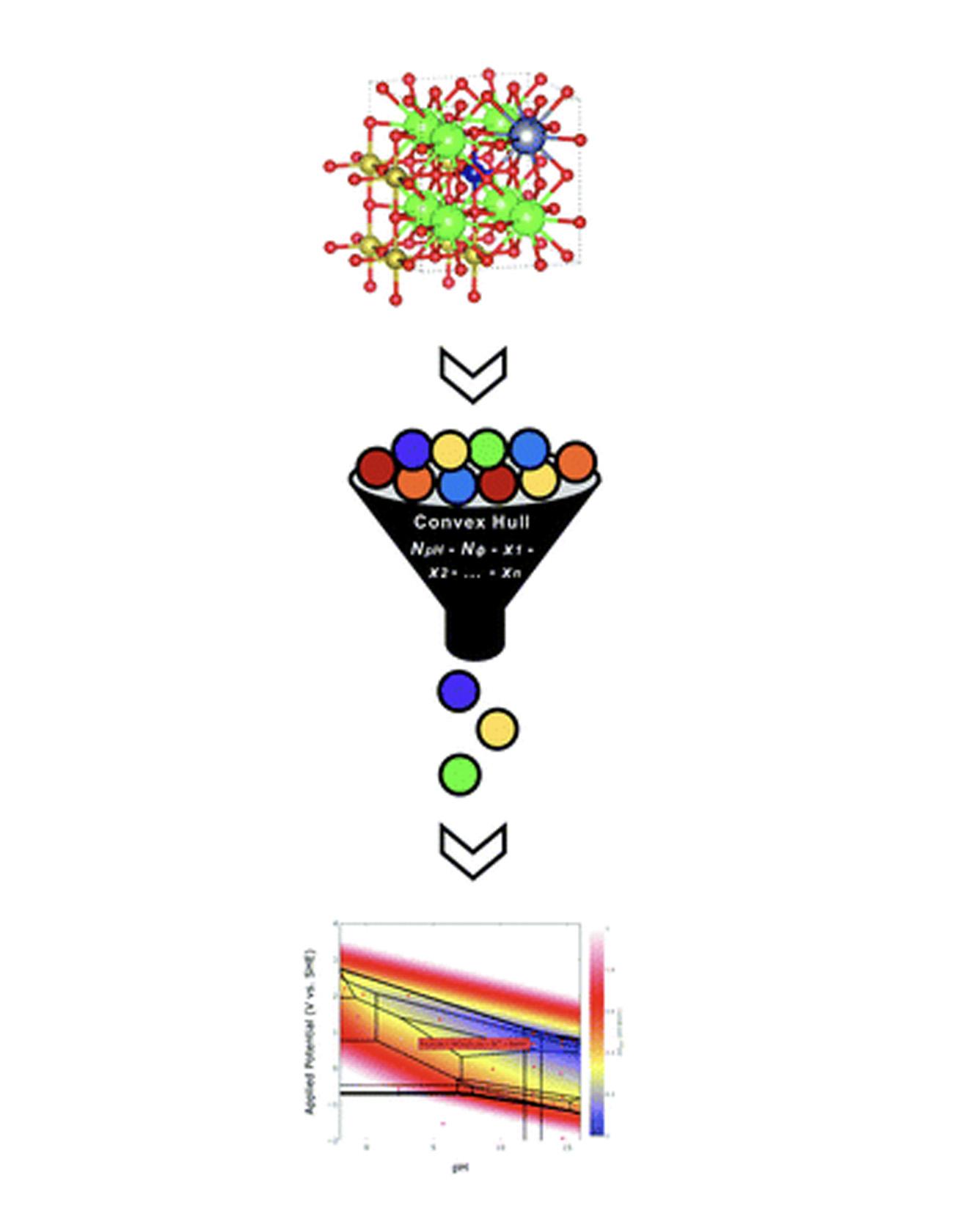
TRI Authors: Muratahan Aykol,Linda Hung, Santosh Suram, Patrick Herring, Jens S. Hummelshoj
All Authors: Muratahan Aykol, Vinay I. Hegde, Linda Hung, Santosh Suram, Patrick Herring, Chris Wolverton, Jens S. Hummelshoj
Assessing the synthesizability of inorganic materials is a grand challenge for accelerating their discovery using computations. Synthesis of a material is a complex process that depends not only on its thermodynamic stability with respect to others, but also on factors from kinetics, to advances in synthesis techniques, to the availability of precursors. This complexity makes the development of a general theory or first-principles approach to synthesizability currently impractical. Here we show how an alternative pathway to predicting synthesizability emerges from the dynamics of the materials stability network: a scale-free network constructed by combining the convex free-energy surface of inorganic materials computed by high-throughput density functional theory and their experimental discovery timelines extracted from citations. The time-evolution of the underlying network properties allows us to use machine-learning to predict the likelihood that hypothetical, computer-generated materials will be amenable to successful experimental synthesis.
Citation: Aykol, Muratahan, Vinay I. Hegde, Linda Hung, Santosh Suram, Patrick Herring, Chris Wolverton, and Jens S. Hummelshøj. "Network analysis of synthesizable materials discovery." Nature communications 10, no. 1 (2019): 1-7.

TRI Author: Santosh Suram
All Authors: Carla P. Gomes, Junwen Bai, Yexiang Xue, Johan Björck, Brendan Rappazzo, Sebastian Ament, Richard Bernstein, Shufeng Kong, Santosh K. Suram, R. Bruce van Dover, and John M. Gregoire
We introduce CRYSTAL, a multi-agent AI system for crystal-structure phase mapping. CRYSTAL is the first system that can automatically generate a portfolio of physically meaningful phase diagrams for expert-user exploration and selection. CRYSTAL outperforms previous methods to solve the example Pd-Rh-Ta phase diagram, enabling the discovery of a mixed-intermetallic methanol oxidation electrocatalyst. The integration of multiple data-knowledge sources and learning and reasoning algorithms, combined with the exploitation of problem decompositions, relaxations, and parallelism, empowers AI to supersede human scientific data interpretation capabilities and enable otherwise inaccessible scientific discovery in materials science and beyond. Read More
Citation: Gomes, Carla P., Junwen Bai, Yexiang Xue, Johan Björck, Brendan Rappazzo, Sebastian Ament, Richard Bernstein et al. "CRYSTAL: a multi-agent AI system for automated mapping of materials' crystal structures." MRS Communications 9, no. 2 (2019): 600-608.

TRI Author: Jens Hummelshøj
All Authors: Paul C Jennings, Steen Lysgaard, Jens Strabo Hummelshøj, Tejs Vegge, Thomas Bligaard
Materials discovery is increasingly being impelled by machine learning methods that rely on pre-existing datasets. Where datasets are lacking, unbiased data generation can be achieved with genetic algorithms. Here a machine learning model is trained on-the-fly as a computationally inexpensive energy predictor before analyzing how to augment convergence in genetic algorithm-based approaches by using the model as a surrogate. This leads to a machine learning accelerated genetic algorithm combining robust qualities of the genetic algorithm with rapid machine learning. The approach is used to search for stable, compositionally variant, geometrically similar nanoparticle alloys to illustrate its capability for accelerated materials discovery, e.g., nanoalloy catalysts. The machine learning accelerated approach, in this case, yields a 50-fold reduction in the number of required energy calculations compared to a traditional “brute force” genetic algorithm. This makes searching through the space of all homotops and compositions of a binary alloy particle in a given structure feasible, using density functional theory calculations. Read More
Citation: Jennings, Paul C., Steen Lysgaard, Jens Strabo Hummelshøj, Tejs Vegge, and Thomas Bligaard. "Genetic algorithms for computational materials discovery accelerated by machine learning." npj Computational Materials 5, no. 1 (2019): 1-6.
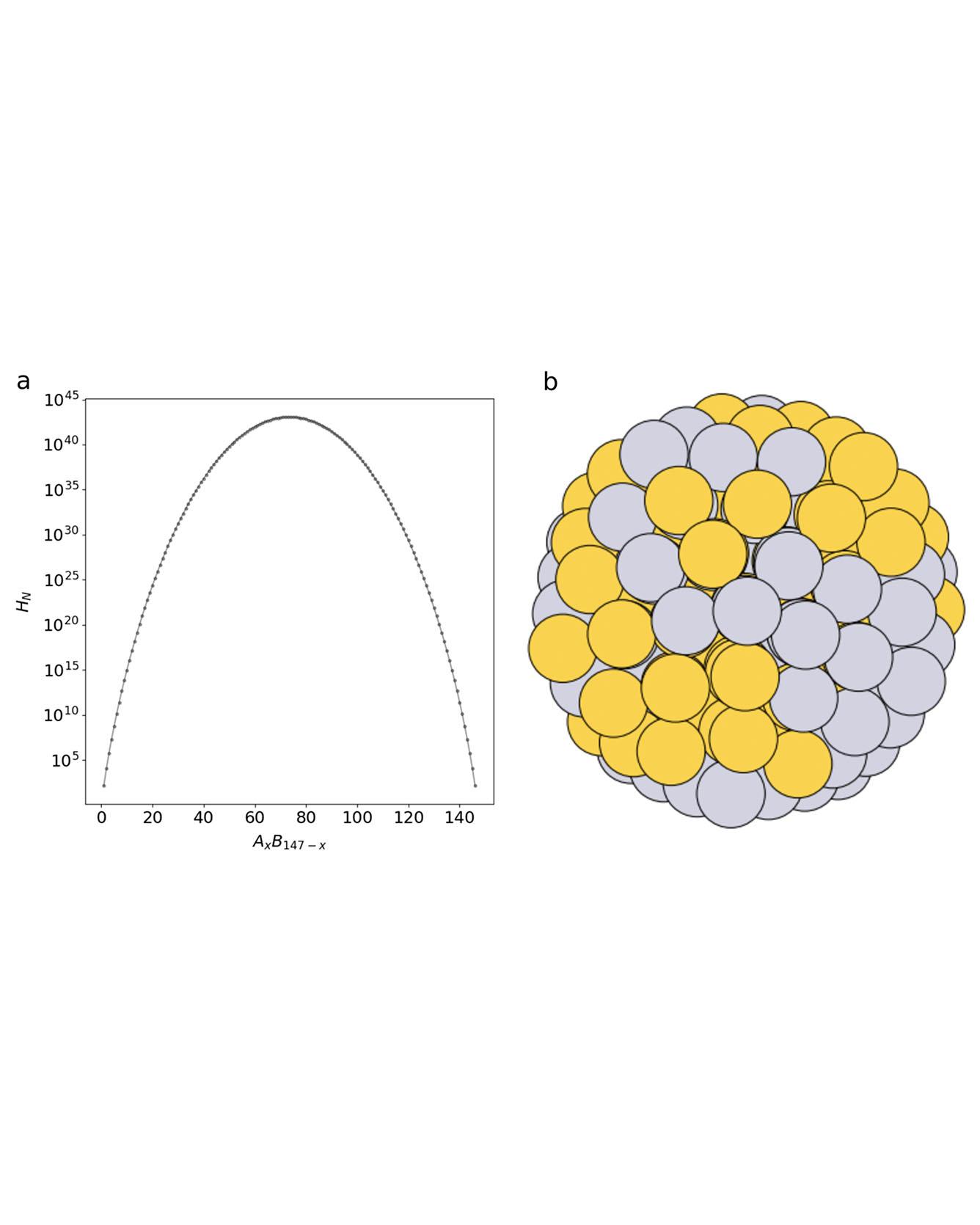
TRI Authors: Muratahan Aykol, Patrick K. Herring
All Authors: Kristen A. Severson, Peter M. Attia, Norman Jin, Nicholas Perkins, Benben Jiang, Zi Yang, Michael H. Chen, Muratahan Aykol, Patrick K. Herring, Dimitrios Fraggedakis, Martin Z. Bazant, Stephen J. Harris, William C. Chueh & Richard D. Braatz
Accurately predicting the lifetime of complex, nonlinear systems such as lithium-ion batteries is critical for accelerating technology development. However, diverse aging mechanisms, significant device variability and dynamic operating conditions have remained major challenges. We generate a comprehensive dataset consisting of 124 commercial lithium iron phosphate/graphite cells cycled under fast-charging conditions, with widely varying cycle lives ranging from 150 to 2,300 cycles. Using discharge voltage curves from early cycles yet to exhibit capacity degradation, we apply machine-learning tools to both predict and classify cells by cycle life. Our best models achieve 9.1% test error for quantitatively predicting cycle life using the first 100 cycles (exhibiting a median increase of 0.2% from initial capacity) and 4.9% test error using the first 5 cycles for classifying cycle life into two groups. This work highlights the promise of combining deliberate data generation with data-driven modelling to predict the behaviour of complex dynamical systems. Read More
Citation: Severson, Kristen A., Peter M. Attia, Norman Jin, Nicholas Perkins, Benben Jiang, Zi Yang, Michael H. Chen et al. "Data-driven prediction of battery cycle life before capacity degradation." Nature Energy 4, no. 5 (2019): 383-391.
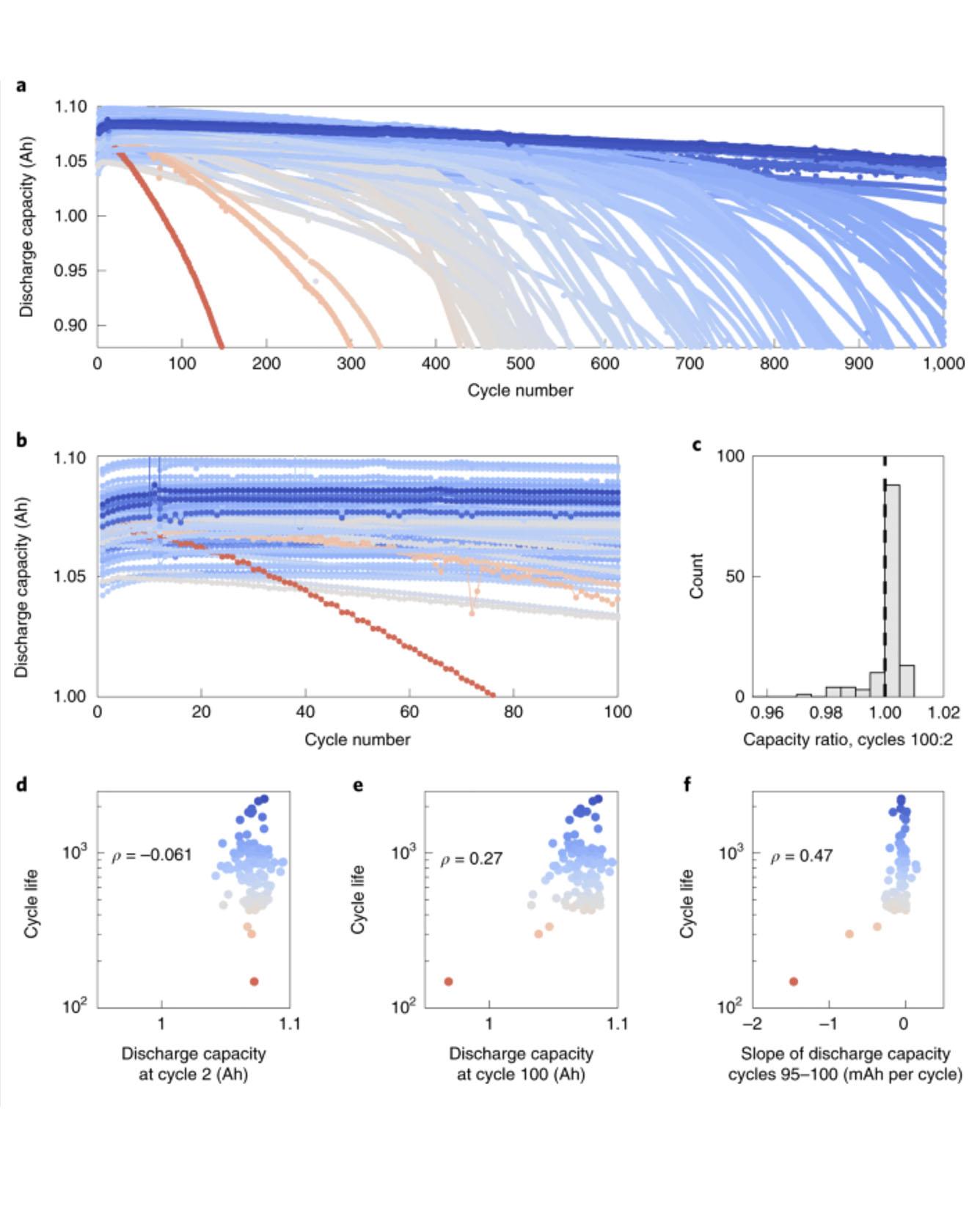
TRI Author: Jens Hummelshøj
All Authors: Steen Lysgaard, Paul C Jennings, Jens Strabo Hummelshøj, Thomas Bligaard, Tejs Vegge
A machine learning model is used as a surrogate fitness evaluator in a genetic algorithm (GA) optimization of the atomic distribution of Pt-Au nanoparticles. The machine learning accelerated genetic algorithm (MLaGA) yields a 50-fold reduction of required energy calculations compared to a traditional GA. Read More
Citation: Lysgaard, Steen, Paul C. Jennings, Jens Strabo Hummelshøj, Thomas Bligaard, and Tejs Vegge. "Machine Learning Accelerated Genetic Algorithms for Computational Materials Search." In ChemRxiv(2018).
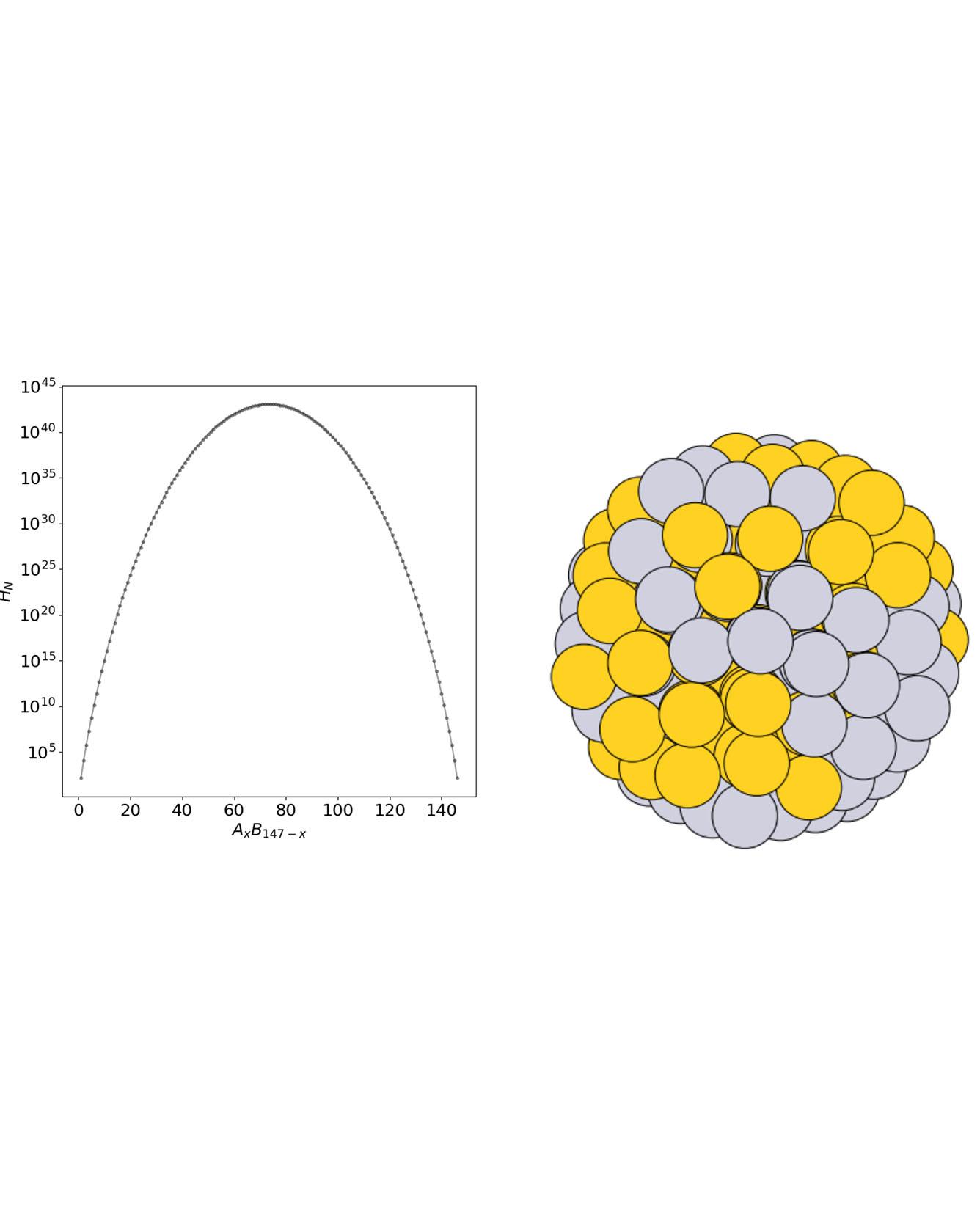
TRI Authors: Muratahan Aykol, Santosh Suram
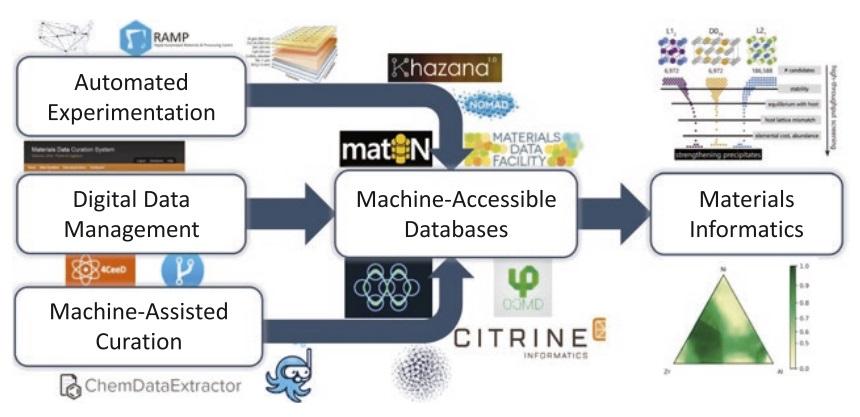
All Authors: Logan Ward, Muratahan Aykol, Ben Blaiszik, Ian Foster, Bryce Meredig, James Saal, Santosh Suram Ongoing, rapid innovations in fields ranging from microelectronics, aerospace, and automotive to defense, energy, and health demand new advanced materials at even greater rates and lower costs. Traditional materials R&D methods offer few paths to achieve both outcomes simultaneously. Materials informatics, while a nascent field, offers such a promise through screening, growing databases of materials for new applications, learning new relationships from existing data resources, and building fast predictive models. We highlight key materials informatics successes from the atomic-scale modeling community, and discuss the ecosystem of open data, software, services, and infrastructure that have led to broad adoption of materials informatics approaches. We then examine emerging opportunities for informatics in materials science and describe an ideal data ecosystem capable of supporting similar widespread adoption of materials informatics, which we believe will enable the faster design of materials. Read More
Citation: Ward, Logan, Muratahan Aykol, Ben Blaiszik, Ian Foster, Bryce Meredig, James Saal, and Santosh Suram. "Strategies for accelerating the adoption of materials informatics." MRS Bulletin 43, no. 9 (2018): 683-689.
TRI Author: Muratahan Aykol
All Authors: Vinay I Hegde, Muratahan Aykol, Scott Kirklin, Chris Wolverton
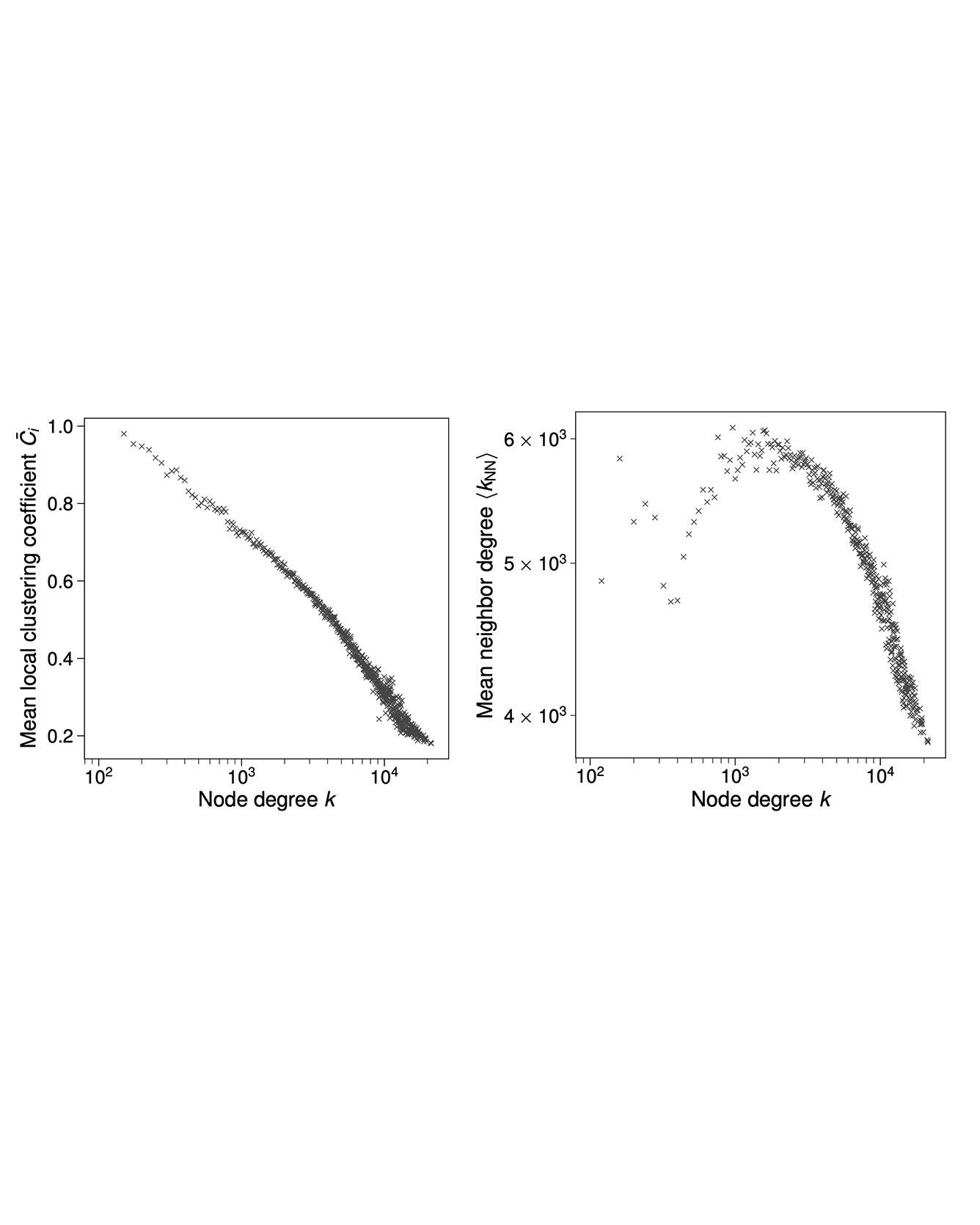
Understanding how the arrangement of atoms and their interactions determine material behavior has been the dominant paradigm in materials science. A complementary approach is studying the organizational structure of networks of materials, defined on the basis of interactions between materials themselves. In this work, we present the "phase diagram of all known inorganic materials", an extremely-dense complex network of nearly 2.1×104 stable inorganic materials (nodes) connected with 41×106 tie-lines (edges) defining their two-phase equilibria, as computed via high-throughput density functional theory. We show that the degree distribution of this network follows a lognormal form, with each material connected to on average 18% of the other materials in the network via tie-lines. Analyzing the structure and topology of this network has potential to uncover new materials knowledge inaccessible from the traditional bottom-up (atoms to materials) approaches. As an example, we derive a data-driven metric for the reactivity of a material as characterized by its connectedness in the network, and quantitatively identify the noblest materials in nature. Read more
Citation: Hegde, Vinay I., Muratahan Aykol, Scott Kirklin, and Chris Wolverton. "The phase diagram of all inorganic materials." In Science Advances, arXiv preprint arXiv:1808.10869 (2018).
TRI Author: Muratahan Aykol
All Authors: Daniel P Tabor, Loïc M Roch, Semion K Saikin, Christoph Kreisbeck, Dennis Sheberla, Joseph H Montoya, Shyam Dwaraknath, Muratahan Aykol, Carlos Ortiz, Hermann Tribukait, Carlos Amador-Bedolla, Christoph J Brabec, Benji Maruyama, Kristin A Persson, Alán Aspuru-Guzik
The discovery and development of novel materials in the field of energy are essential to accelerate the transition to a low-carbon economy. Bringing recent technological innovations in automation, robotics and computer science together with current approaches in chemistry, materials synthesis and characterization will act as a catalyst for revolutionizing traditional research and development in both industry and academia. This Perspective provides a vision for an integrated artificial intelligence approach towards autonomous materials discovery, which, in our opinion, will emerge within the next 5 to 10 years. The approach we discuss requires the integration of the following tools, which have already seen substantial development to date: high-throughput virtual screening, automated synthesis planning, automated laboratories and machine learning algorithms. In addition to reducing the time to deployment of new materials by an order of magnitude, this integrated approach is expected to lower the cost associated with the initial discovery. Thus, the price of the final products (for example, solar panels, batteries and electric vehicles) will also decrease. This in turn will enable industries and governments to meet more ambitious targets in terms of reducing greenhouse gas emissions at a faster pace. Read More
Citation: Tabor, Daniel P., Loïc M. Roch, Semion K. Saikin, Christoph Kreisbeck, Dennis Sheberla, Joseph H. Montoya, Shyam Dwaraknath et al. "Accelerating the discovery of materials for clean energy in the era of smart automation." Nature Reviews Materials 3, no. 5 (2018): 5-20.