Featured Publications

All Publications
The Open Databases Integration for Materials Design (OPTIMADE) consortium has designed a universal application programming interface (API) to make materials databases accessible and interoperable. We outline the first stable release of the specification, v1.0, which is already supported by many leading databases and several software packages. We illustrate the advantages of the OPTIMADE API through worked examples on each of the public materials databases that support the full API specification. READ MORE
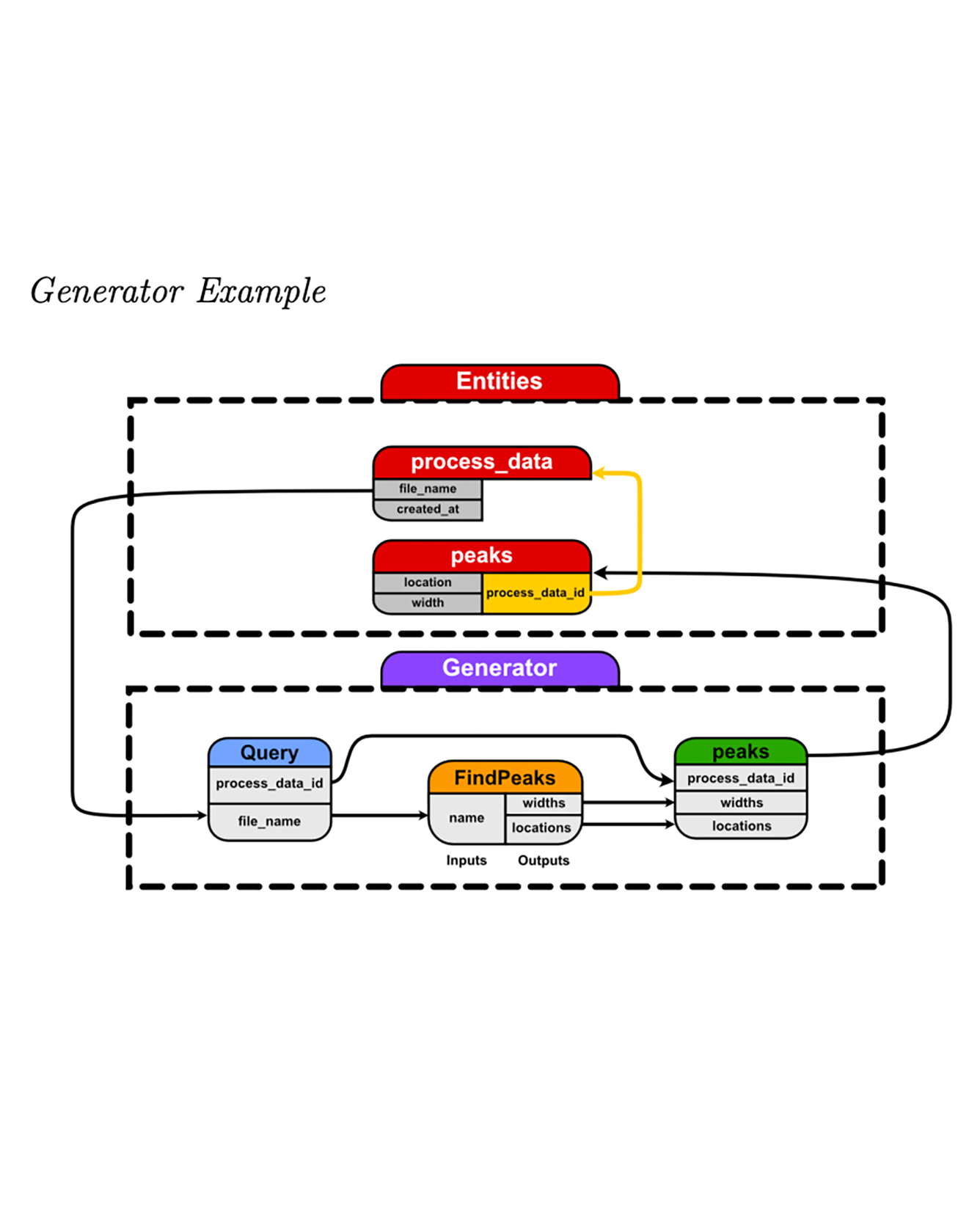
In this work, we present DBgen, a Python library that provides a framework for defining extract-transform-load (ETL) pipelines to create and populate SQL databases. DBgen is most useful when the underlying data has complex relationships, requires multi-step analysis, is large-scale, and the type of data being collected changes frequently. Scientific data often fits this description. With current tooling, defining ETL pipelines for this particularly difficult- to-manage data is so onerous that a great deal of it does not end up being stored in a database and is opaque. DBgen is designed to fill the gap in the current tooling and reduce the barrier to defining ETL pipelines such data. READ MORE
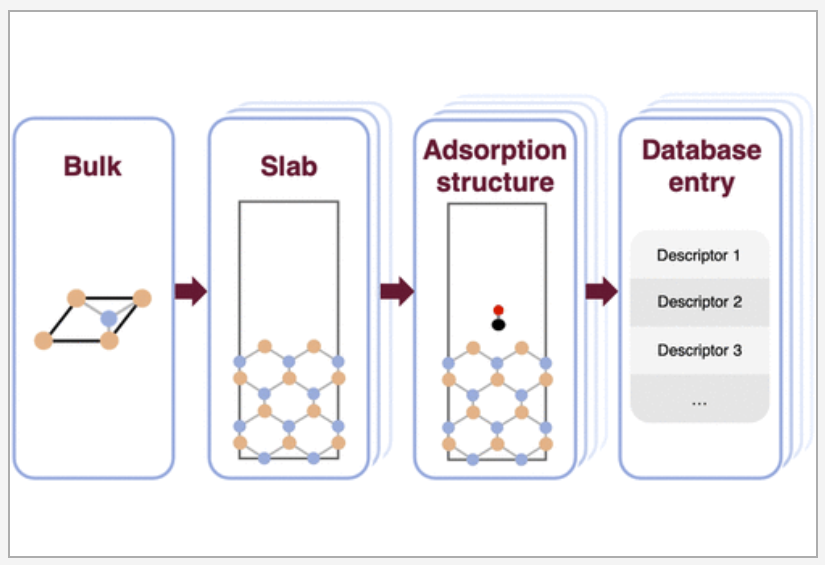
Surface adsorption is a crucial step in numerous processes, including heterogeneous catalysis, where the adsorption of key species is often used as a descriptor of efficiency. We present here an automated adsorption workflow for semiconductors which employs density functional theory calculations to generate adsorption data in a high-throughput manner. Starting from a bulk structure, the workflow performs an exhaustive surface search, followed by an adsorption structure construction step, which generates a minimal energy landscape to determine the optimal adsorbate–surface distance. An extensive set of energy-based, charge-based, geometric, and electronic descriptors tailored toward catalysis research are computed and saved to a personal user database. The application of the workflow to zinc telluride, a promising CO2 reduction photocatalyst, is presented as a case study to illustrate the capabilities of this method and its potential as a material discovery tool. READ MORE
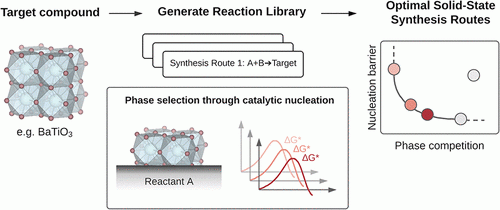
The rational solid-state synthesis of inorganic compounds is formulated as catalytic nucleation on crystalline reactants, where contributions of reaction and interfacial energies to the nucleation barriers are approximated from high-throughput thermochemical data and structural and interfacial features of crystals, respectively. Favorable synthesis reactions are then identified by a Pareto analysis of relative nucleation barriers and phase selectivities of reactions leading to the target. We demonstrate the application of this approach in reaction planning for the solid-state synthesis of a range of compounds, including the widely studied oxides LiCoO2, BaTiO3, and YBa2Cu3O7, as well as other metal oxide, oxyfluoride, phosphate, and nitride targets. Pathways for enabling the retrosynthesis of inorganics are also discussed. READ MORE
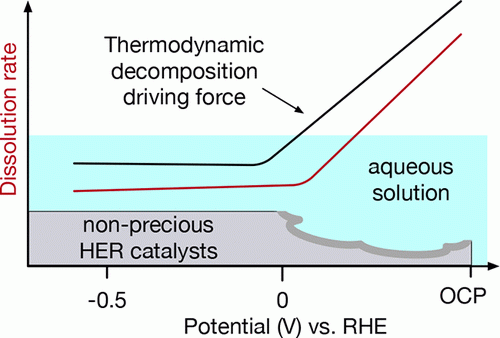
Nonprecious hydrogen evolution reaction (HER) catalysts commonly suffer from severe dissolution under open-circuit potential (OCP). In this work, using calculated Pourbaix diagrams, we quantitatively analyze the stability of a set of well-known active HER catalysts (MoS2, MoP, CoP, Pt in acid, and Ni3Mo in base) under working conditions. We determine that the large thermodynamic driving force toward decomposition created by the electrode/electrolyte interface potential is responsible for the substantial dissolution of nonprecious HER catalysts at OCP. Our analysis further shows the stability of HER catalysts in acidic solution is ordered as Pt ≈ MoS2 > MoP > CoP, which is confirmed by the measured dissolution rates using an inductively coupled plasma mass spectrometer. On the basis of the gained insights, we suggest strategies to circumvent the catalyst dissolution in aqueous solution. READ MORE
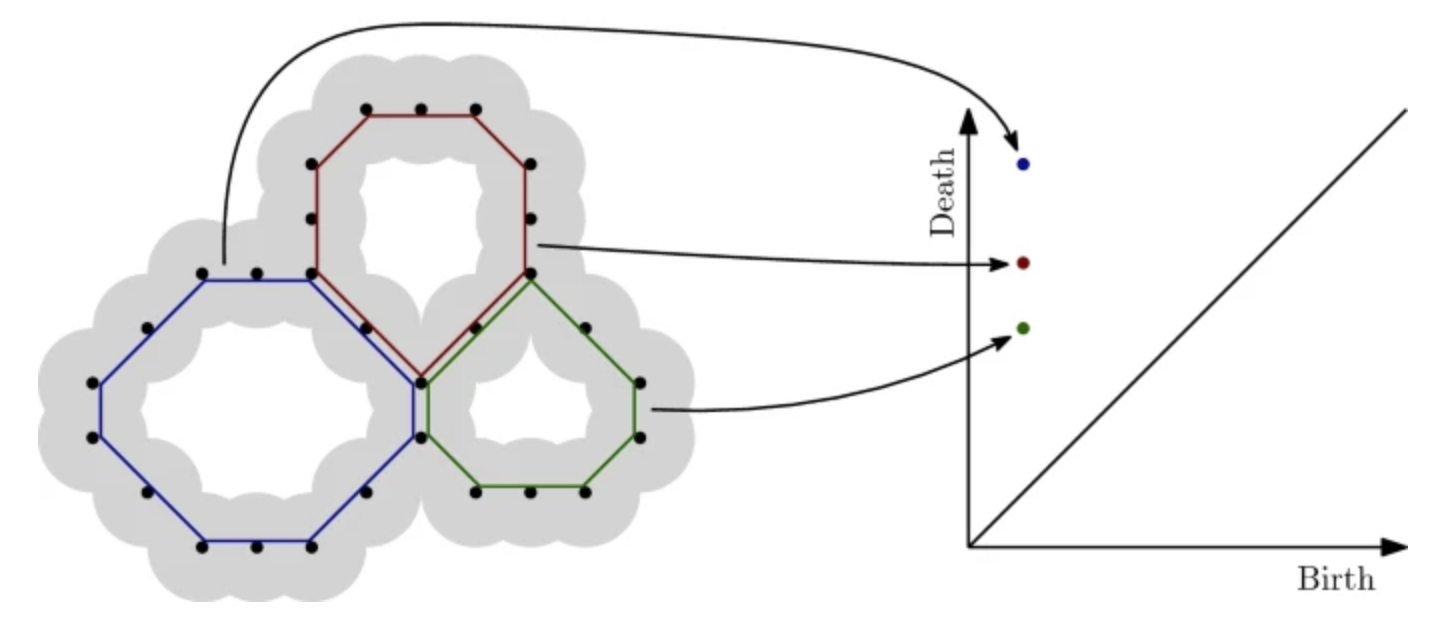
Machine learning has emerged as a powerful approach in materials discovery. Its major challenge is selecting features that create interpretable representations of materials, useful across multiple prediction tasks. We introduce an end-to-end machine learning model that automatically generates descriptors that capture a complex representation of a material’s structure and chemistry. This approach builds on computational topology techniques (namely, persistent homology) and word embeddings from natural language processing. It automatically encapsulates geometric and chemical information directly from the material system. We demonstrate our approach on multiple nanoporous metal–organic framework datasets by predicting methane and carbon dioxide adsorption across different conditions. Our results show considerable improvement in both accuracy and transferability across targets compared to models constructed from the commonly-used, manually-curated features, consistently achieving an average 25–30% decrease in root-mean-squared-deviation and an average increase of 40–50% in R2 scores. A key advantage of our approach is interpretability: Our model identifies the pores that correlate best to adsorption at different pressures, which contributes to understanding atomic-level structure–property relationships for materials design. READ MORE
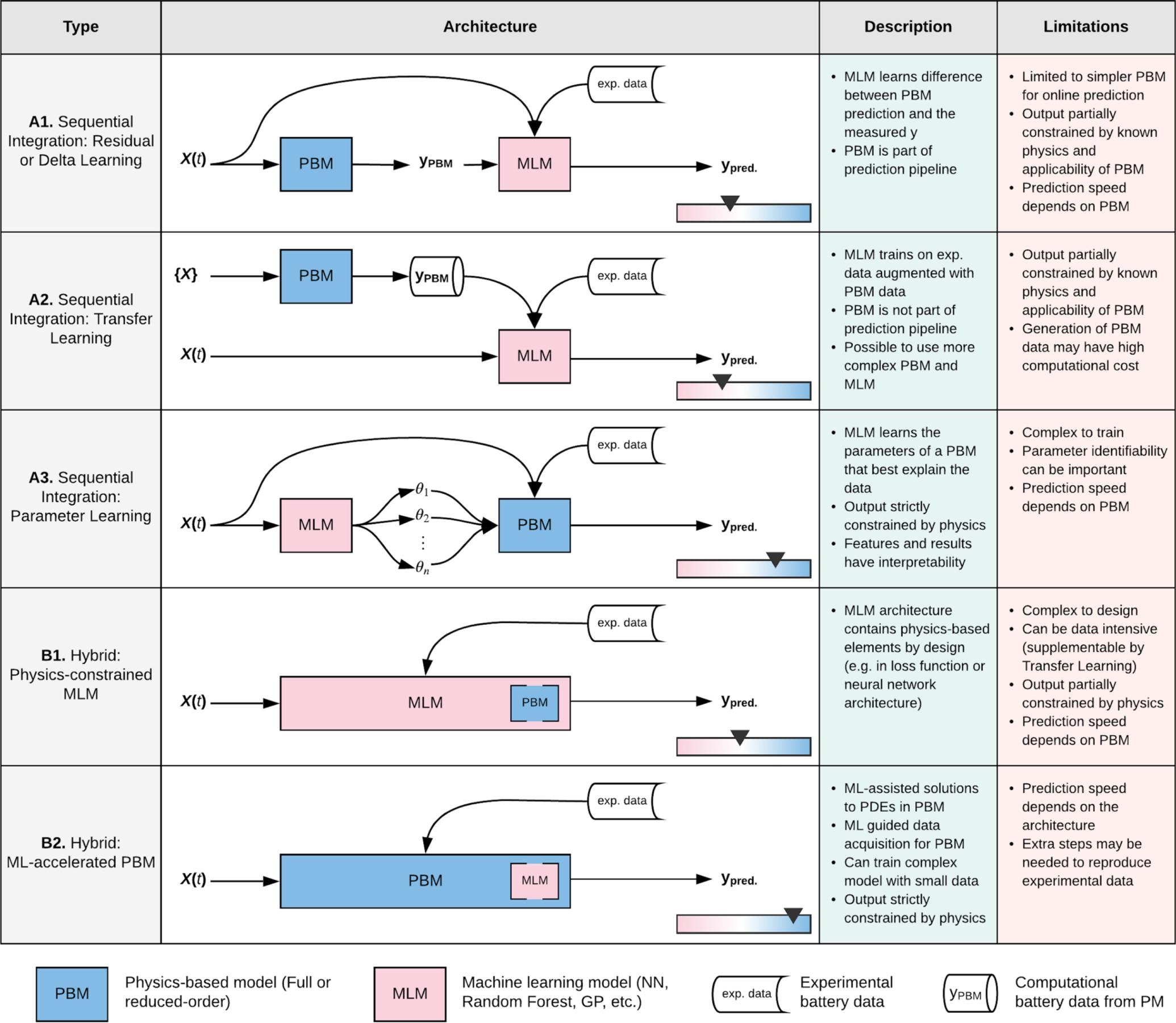
Forecasting the health of a battery is a modeling effort that is critical to driving improvements in and adoption of electric vehicles. Purely physics-based models and purely data-driven models have advantages and limitations of their own. Considering the nature of battery data and end-user applications, we outline several architectures for integrating physics-based and machine learning models that can improve our ability to forecast battery lifetime. We discuss the ease of implementation, advantages, limitations, and viability of each architecture, given the state of the art in the battery and machine learning fields. READ MORE
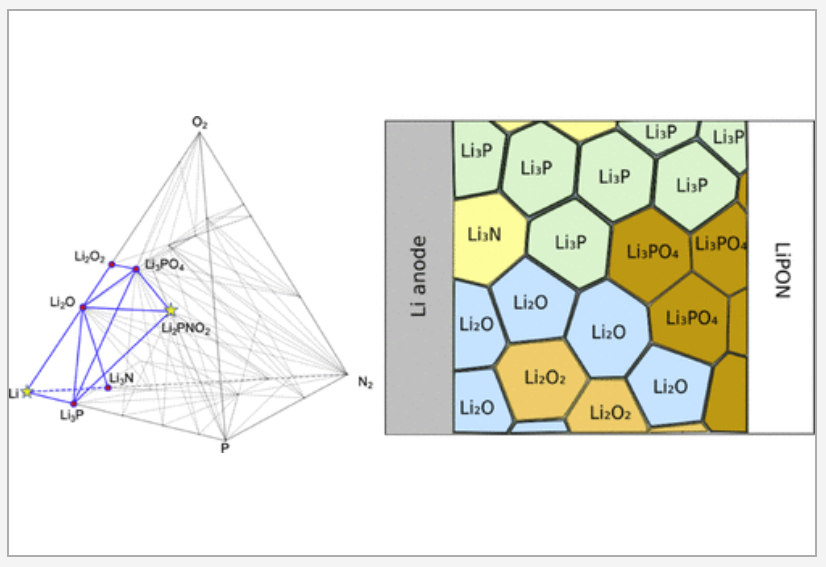
All-solid-state lithium-ion batteries have attracted significant research interest for providing high power and energy densities with enhanced operational safety. Despite the discoveries of solid electrolyte materials with superionic conductivities, it remains a challenge to maintain high rate capability in all-solid lithium-ion batteries in long-term operation. The observed rate degradation has been attributed to reactivity and resistance at the electrode–electrolyte interfaces. We examine interfaces formed between eight electrolytes including garnet, LiPON, and Li10GeP2S12 (LGPS) and seven electrode materials including an NCM cathode and a metallic Li anode and identify the most rapid lithium-ion diffusion pathways through metastable arrangements of product phases that may precipitate out at each interface. Our analysis accounts for possible density functional theory (DFT) error, metastability, and finite-temperature effects by statistically sampling thousands of possible phase diagrams for each interface. The lithium-ion conductivities in the product phases at the interface are evaluated using machine-learned interatomic potentials trained on the fly. In nearly all electrode–electrolyte interfaces we evaluate, we predict that lithium-ion conduction in the product phases making up the interphase region becomes the rate-limiting step for battery performance. READ MORE
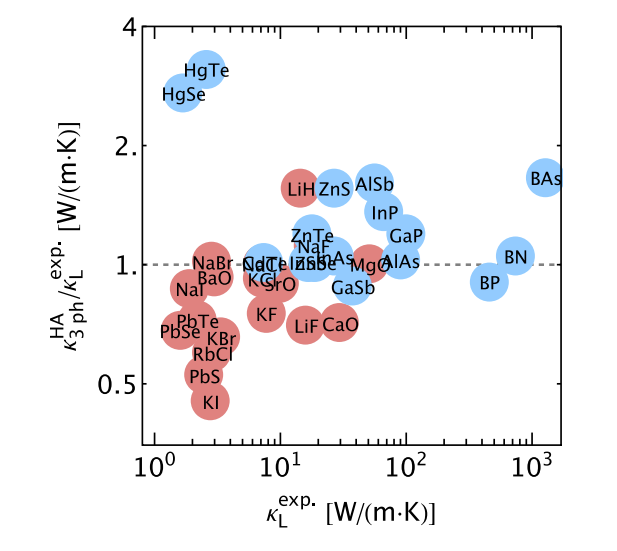
Thermal transport phenomena are ubiquitous and play a critical role in the performance of various microelectronic and energy-conversion devices. Binary rocksalt and zinc blende compounds, despite their rather simple crystal structures, exhibit an extraordinary range of lattice thermal conductivity (κL) spanning over 3 orders of magnitude. A comprehensive understanding of the underlying heat transfer mechanism through the development of microscopic theories is therefore of fundamental importance, yet it remains elusive because of the challenges arising from explicitly treating higher-order anharmonicity. Recent theoretical and experimental advances have revealed the essential role of quartic anharmonicity in suppressing heat transfer in zinc blende boron arsenide (BAs) with ultrahigh κL. However, critical questions concerning the general effects of higher-order anharmonicity in the broad classes and chemistries of binary solids are still unanswered. Using our recently developed high-throughput phonon framework based on first-principles density functional theory calculations, we systematically investigate the lattice dynamics and thermal transport properties of 37 binary compounds with rocksalt and zinc blende structures at room temperature, with a particular focus on unraveling the impacts of quartic anharmonicity on κL. Our advanced theoretical model for computing κL embraces current state-of-the-art methods, featuring a complete treatment of quartic anharmonicity for both phonon frequencies and lifetimes at finite temperatures, as well as contributions from off-diagonal terms in the heat-flux operator. We find the impacts of quartic anharmonicity on κL to be strikingly different in rocksalt and zinc blende compounds, owing to the countervailing effects on finite-temperature-induced shifts in phonon frequencies and scattering rates. By correlating κL with the phonon scattering phase space, we outline a qualitative but efficient route to assess the importance of four-phonon scattering from harmonic phonon calculations. Among notable examples, in zinc blende HgTe, we identify an unprecedented sixfold reduction in κL due to four-phonon scattering, which dominates over the three-phonon scattering in the acoustic region at room temperature. We also demonstrate a possible breakdown of the phonon gas model in rocksalt AgCl, wherein the phonon states are significantly broadened due to strong intrinsic anharmonicity, inducing off-diagonal contributions to κL comparable to the diagonal ones. The deep physical insights gained in this work can be used to guide the rational design of thermal management materials. READ MORE
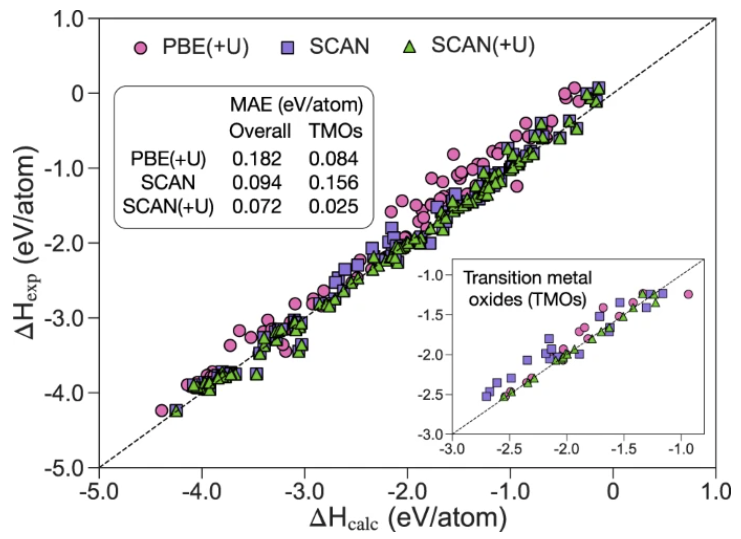
In this work, using the SCAN functional, we develop a simple method on top of the Materials Project (MP) Pourbaix diagram framework to accurately predict the aqueous stability of solids. We extensively evaluate the SCAN functional’s performance in computed formation enthalpies for a broad range of oxides and develop Hubbard U corrections for transition-metal oxides where the standard SCAN functional exhibits large deviations. The performance of the calculated Pourbaix diagram using the SCAN functional is validated with comparison to the experimental and the MP PBE Pourbaix diagrams for representative examples. Benchmarks indicate the SCAN Pourbaix diagram systematically outperforms the MP PBE in aqueous stability prediction. We further show applications of this method in accurately predicting the dissolution potentials of the state-of-the-art catalysts for oxygen evolution reaction in acidic media. READ MORE