Featured Publications

All Publications
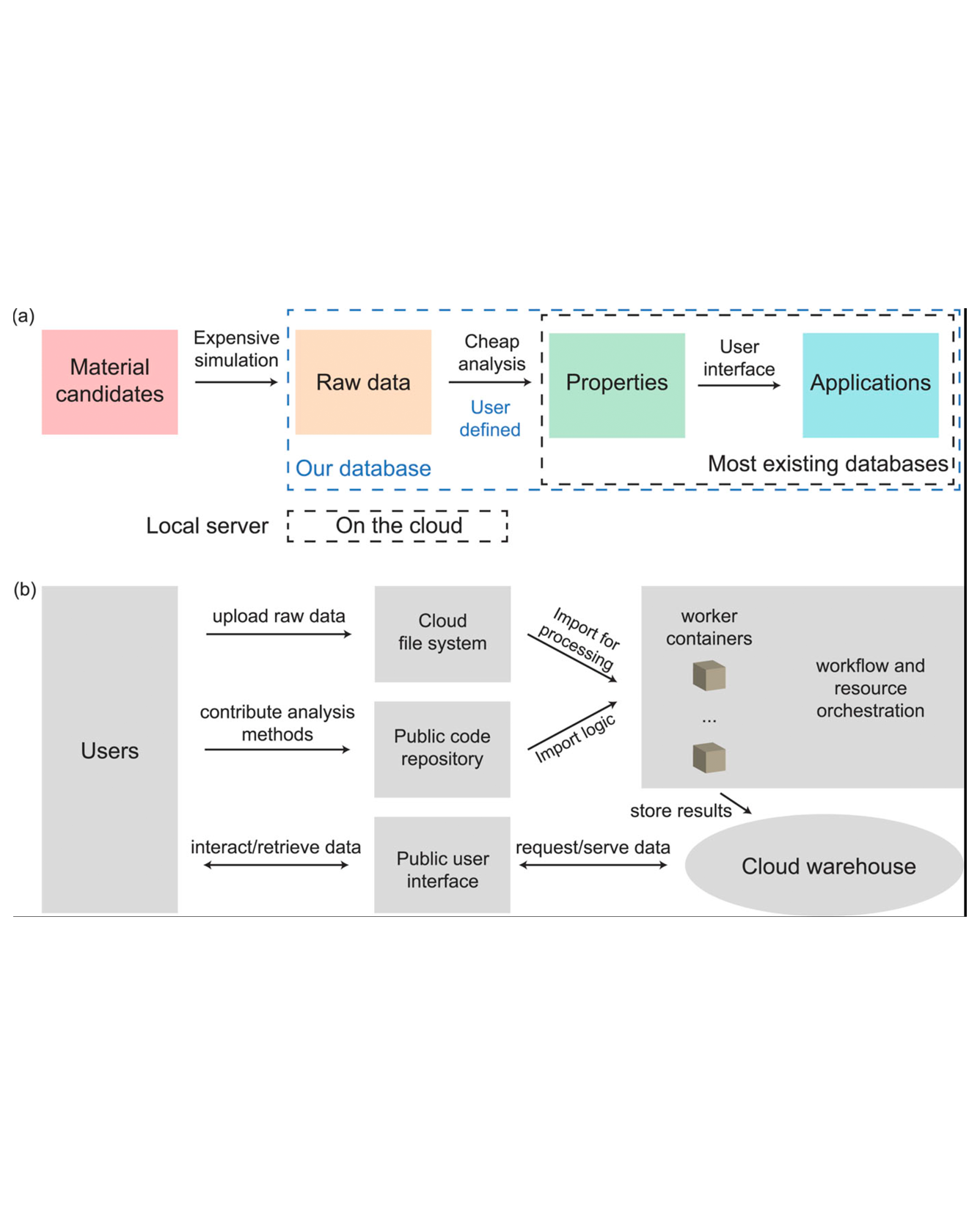
Open material databases storing thousands of material structures and their properties have become the cornerstone of modern computational materials science. Yet, the raw simulation outputs are generally not shared due to their huge size. In this work, we describe a cloud-based platform to enable fast post-processing of the trajectories and to facilitate sharing of the raw data. As an initial demonstration, our database includes 6286 molecular dynamics trajectories for amorphous polymer electrolytes (5.7 terabytes of data). We create a public analysis library at https://github.com/TRI-AMDD/htp_md to extract ion transport properties from the raw data using expert-designed functions and machine learning models. The analysis is run automatically on the cloud, and the results are uploaded onto an open database. Our platform encourages users to contribute both new trajectory data and analysis functions via public interfaces. Finally, we create a front-end user interface at https://www.htpmd.matr.io/ for browsing and visualization of our data. We envision the platform to be a new way of sharing raw data and new insights for the materials science community. READ MORE
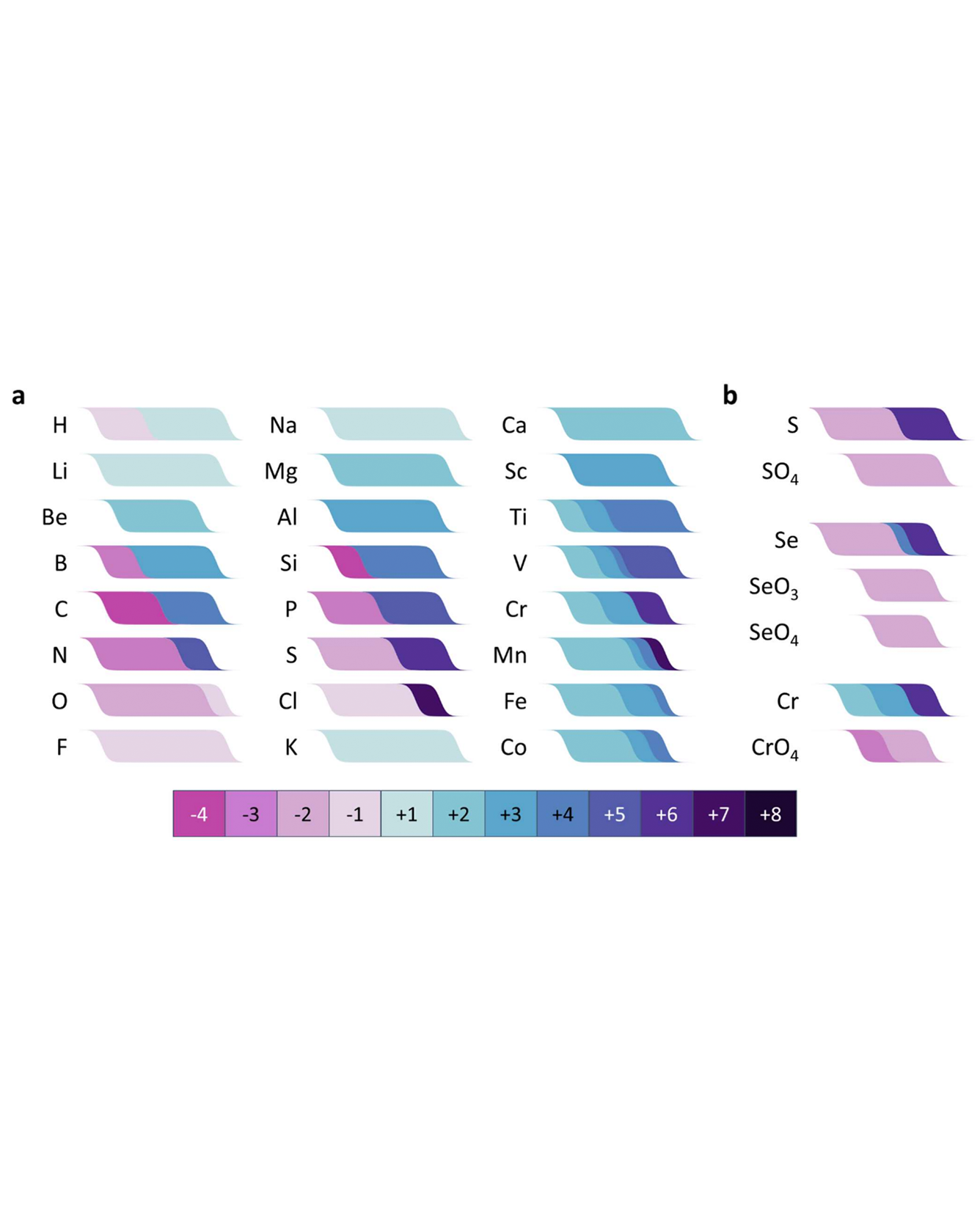
The electrochemical series is a useful tool in electrochemistry, but its effectiveness in materials chemistry is limited by the fact that the standard electrochemical series is based on a relatively small set of reactions, many of which are measured in aqueous solutions. We have used machine learning to create an electrochemical series for inorganic materials from tens of thousands of entries in the Inorganic Crystal Structure Database. We demonstrate that this approach enables the prediction of oxidation states directly from composition in a way that is physically justified, human-interpretable, and more accurate than a state-of-the-art transformer-based neural network model. We present applications of our model to structure prediction, materials discovery, and materials electrochemistry, and we discuss possible additional applications and areas for improvement. To facilitate the use of our approach, we introduce a freely available web site and API. READ MORE
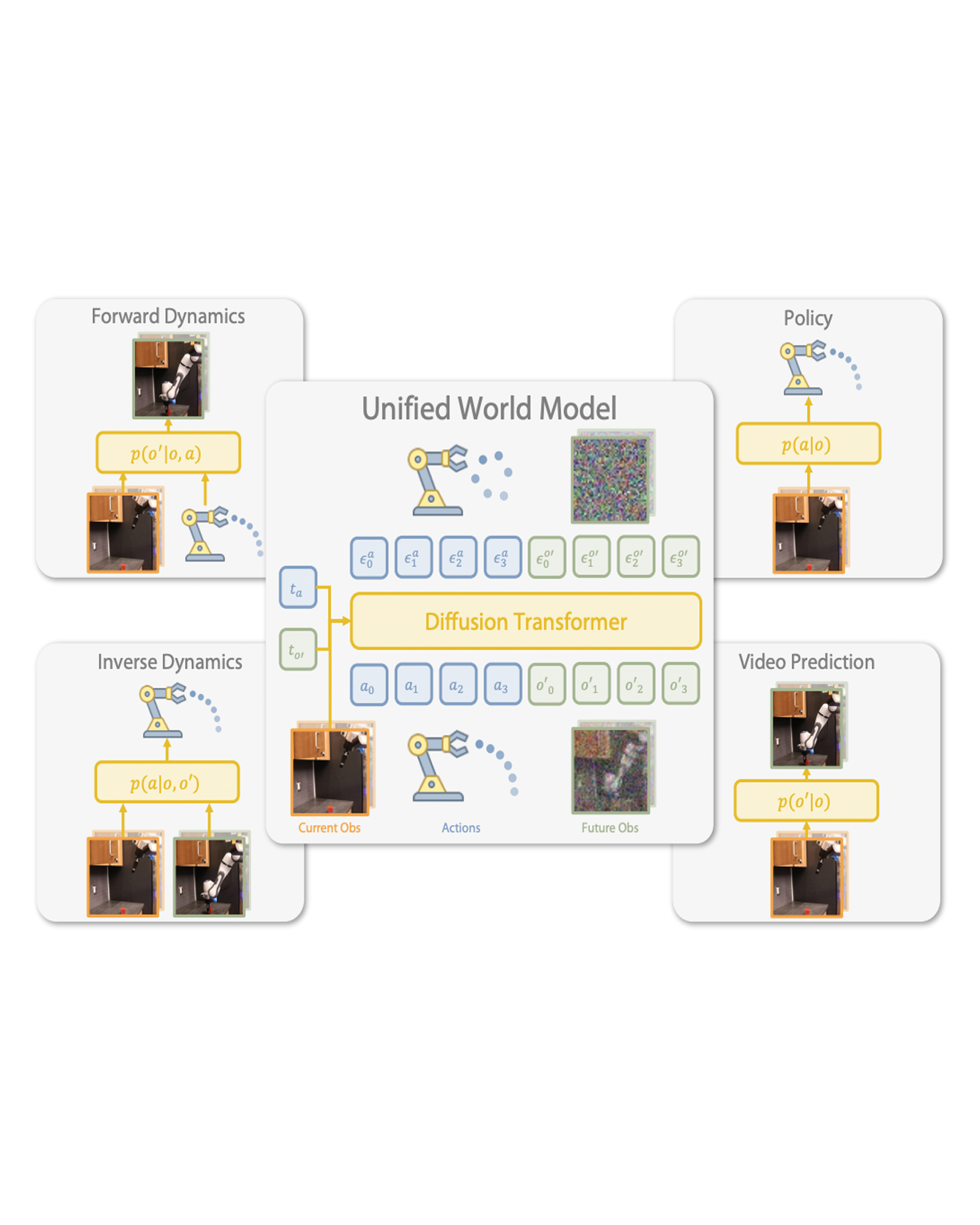
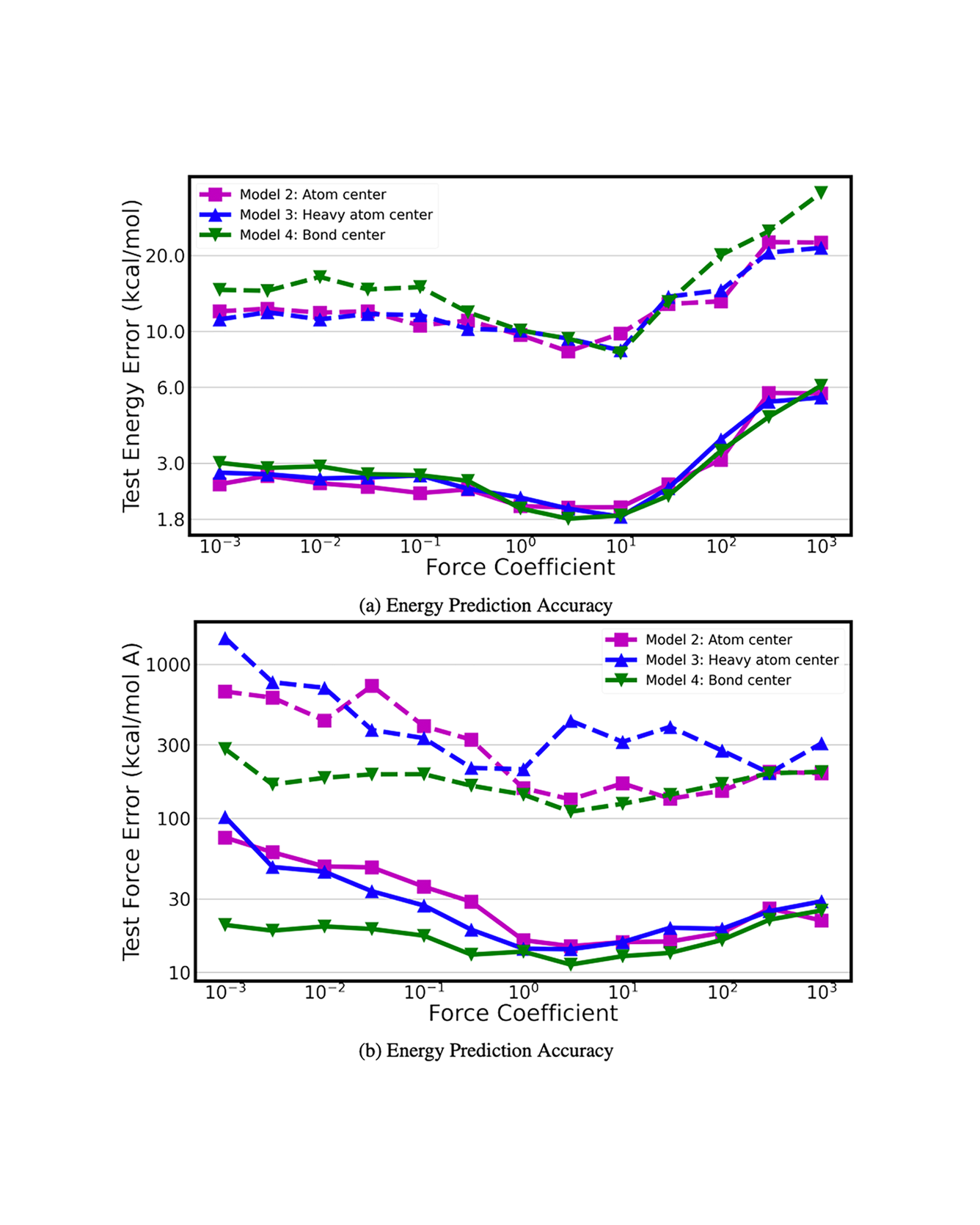
This paper introduces the Chemical Environment Modeling Theory (CEMT), a novel, generalized framework designed to overcome the limitations inherent in traditional atom-centered Machine Learning Force Field (MLFF) models, widely used in atomistic simulations of chemical systems. CEMT demonstrated enhanced flexibility and adaptability by allowing reference points to exist anywhere within the modeled domain and thus, enabling the study of various model architectures. Utilizing Gaussian Multipole (GMP) featurization functions, several models with different reference point sets, including finite difference grid-centered and bond-centered models, were tested to analyze the variance in capabilities intrinsic to models built on distinct reference points. The results underscore the potential of non-atom-centered reference points in force training, revealing variations in prediction accuracy, inference speed and learning efficiency. Finally, a unique connection between CEMT and real-space orbital-free finite element Density Functional Theory (FE-DFT) is established, and the implications include the enhancement of data efficiency and robustness. It allows the leveraging of spatially-resolved energy densities and charge densities from FE-DFT calculations, as well as serving as a pivotal step towards integrating known quantum-mechanical laws into the architecture of ML models. READ MORE
Self-driving labs (SDLs) leverage combinations of artificial intelligence, automation, and advanced computing to accelerate scientific discovery. The promise of this field has given rise to a rich community of passionate scientists, engineers, and social scientists, as evidenced by the development of the Acceleration Consortium and recent Accelerate Conference. Despite its strengths, this rapidly developing field presents numerous opportunities for growth, challenges to overcome, and potential risks of which to remain aware. This community perspective builds on a discourse instantiated during the first Accelerate Conference, and looks to the future of self-driving labs with a tempered optimism. Incorporating input from academia, government, and industry, we briefly describe the current status of self-driving labs, then turn our attention to barriers, opportunities, and a vision for what is possible. Our field is delivering solutions in technology and infrastructure, artificial intelligence and knowledge generation, and education and workforce development. In the spirit of community, we intend for this work to foster discussion and drive best practices as our field grows. READ MORE

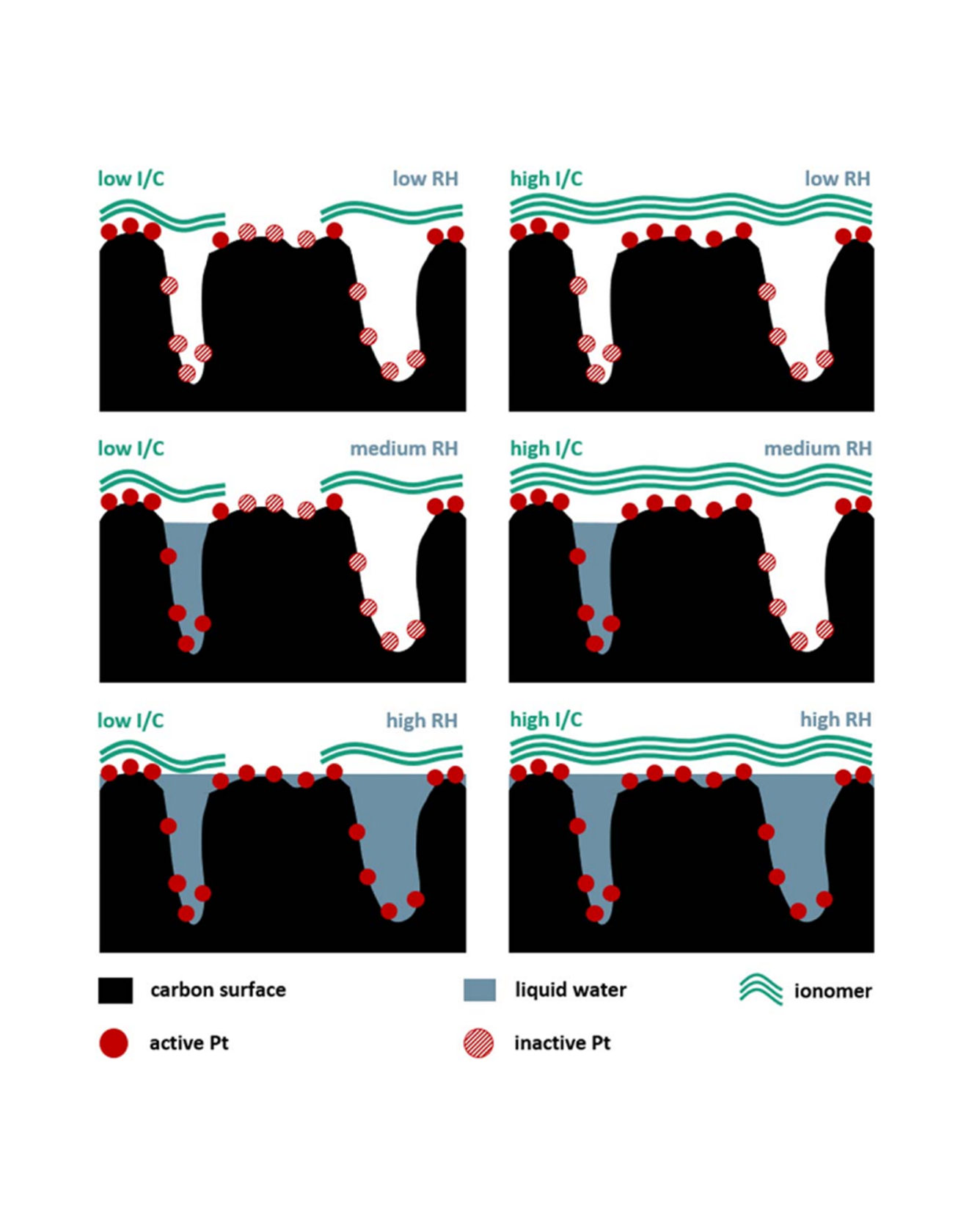
The effect of ionomer to carbon (I/C) weight ratio and relative humidity (RH) on cathode catalyst degradation was investigated by comprehensive in situ characterization. Membrane electrode assemblies (MEA) with I/C ratios of 0.5, 0.8 and 1.2 were subjected to an accelerated stress test performed at 40, 70 and 100% RH. The results show an increasing loss in electrochemical active surface area (ECSA) for both higher I/C ratios and RH during voltage cycling. To differentiate between ionomer and water connected ECSA, carbon monoxide stripping measurements were performed at varying RH. Before degradation, all MEAs show comparable total ECSA values, while higher I/C ratios lead to a larger fraction of ionomer connected ECSA. After degradation, ECSA measurements of the lowest I/C ratio showed a relatively higher loss of Pt in contact with ionomer than Pt in contact with water, while an opposite trend was observed for higher I/C ratios. H2/N2 impedance measurements showed drastically increasing protonic catalyst layer resistances for decreasing RH especially at low I/C ratios, which might hinder Pt2+ ion diffusion towards the membrane, hence decreasing the ECSA loss. Limiting current measurements show increasing molecular O2 diffusion resistances at end of test for samples with higher I/C ratios and higher ECSA loss. READ MORE
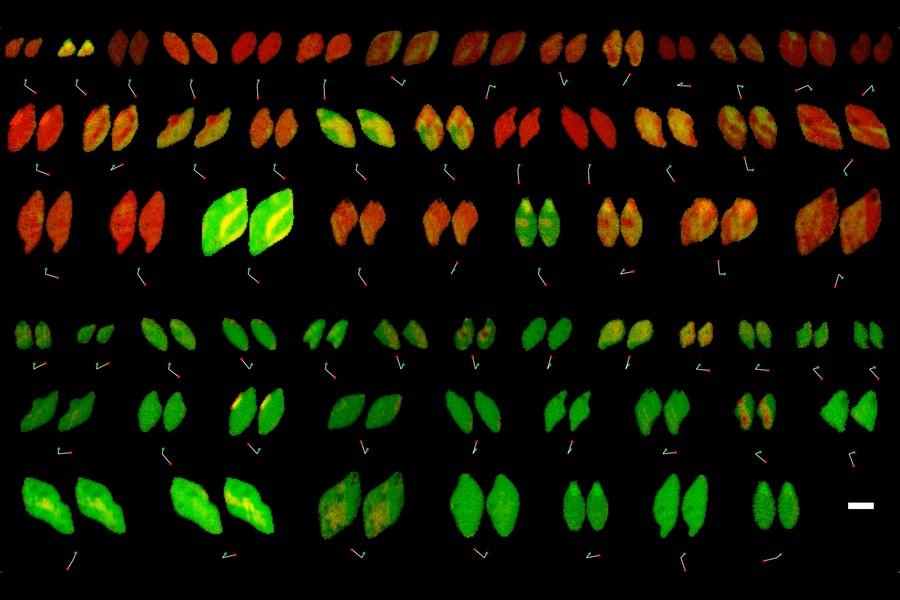
Reaction rates at spatially heterogeneous, unstable interfaces are notoriously difficult to quantify, yet are essential in engineering many chemical systems, such as batteries1 and electrocatalysts2. Experimental characterizations of such materials by operando microscopy produce rich image datasets3,4,5,6, but data-driven methods to learn physics from these images are still lacking because of the complex coupling of reaction kinetics, surface chemistry and phase separation7. Here we show that heterogeneous reaction kinetics can be learned from in situ scanning transmission X-ray microscopy (STXM) images of carbon-coated lithium iron phosphate (LFP) nanoparticles. Combining a large dataset of STXM images with a thermodynamically consistent electrochemical phase-field model, partial differential equation (PDE)-constrained optimization and uncertainty quantification, we extract the free-energy landscape and reaction kinetics and verify their consistency with theoretical models. We also simultaneously learn the spatial heterogeneity of the reaction rate, which closely matches the carbon-coating thickness profiles obtained through Auger electron microscopy (AEM). Across 180,000 image pixels, the mean discrepancy with the learned model is remarkably small (<7%) and comparable with experimental noise. Our results open the possibility of learning nonequilibrium material properties beyond the reach of traditional experimental methods and offer a new non-destructive technique for characterizing and optimizing heterogeneous reactive surfaces. READ MORE
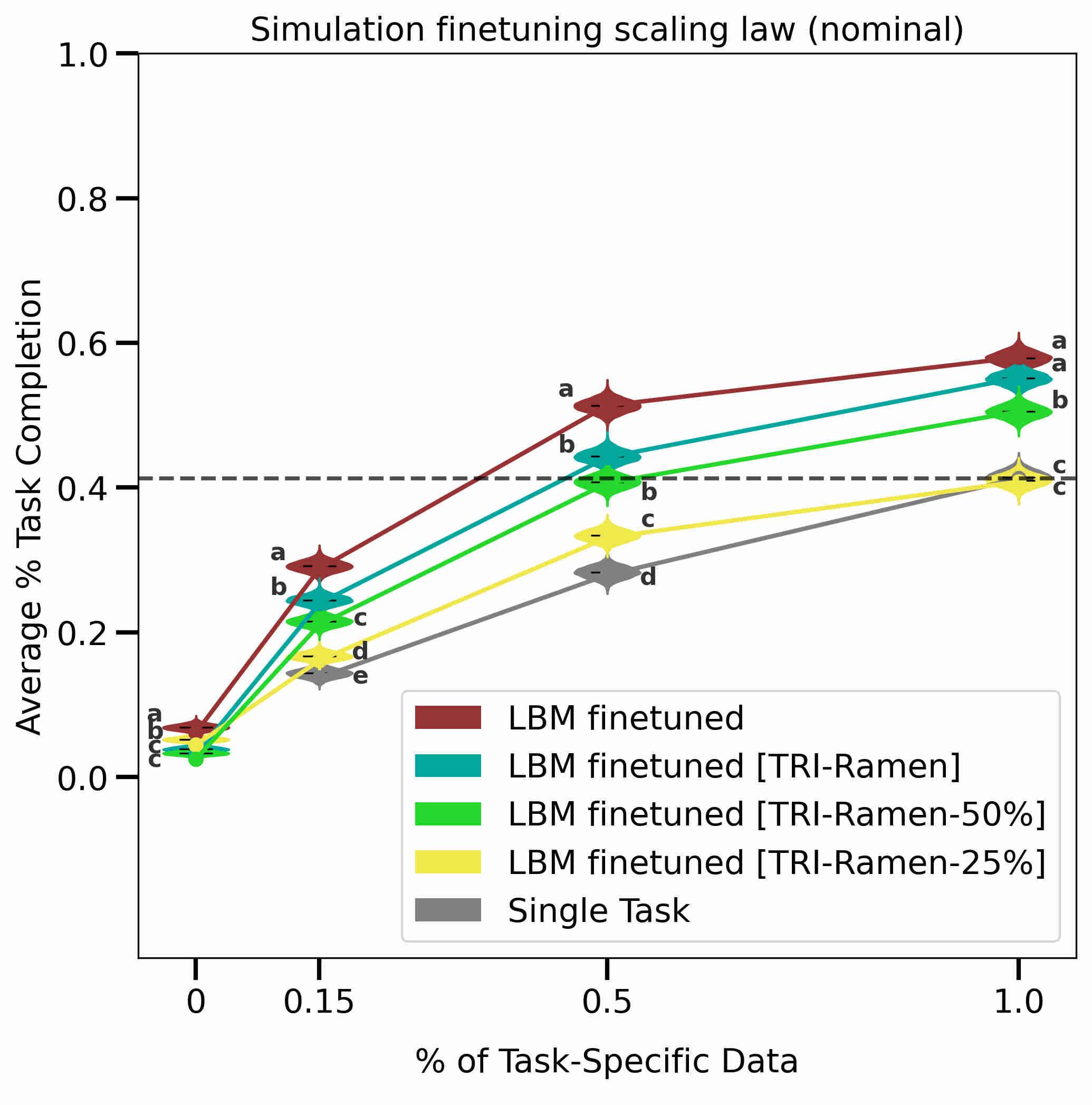
Data-driven models are being developed to predict battery lifetime because of their ability to capture complex aging phenomena. In this perspective, we demonstrate that it is critical to consider the use cases when developing prediction models. Specifically, model features need to be classified to differentiate whether or not they encode cycling conditions, which are sometimes used to artificially increase the diversity in battery lifetime. Many use cases require the prediction of cell-to-cell variability between identically cycled cells, such as production quality control. Developing models for such prediction tasks thus requires features that do not rely on cycling conditions. Using the dataset published by Severson et al. in 2019 as an example, we show that features encoding cycling conditions boost model accuracy because they predict the protocol-to-protocol variability. However, models based on these features are less transferable when deployed on identically cycled cells. Our analysis underscores the concept of using the right features for the right prediction task. We encourage researchers to consider the usage scenarios that they are developing models for and whether or not to include cycling conditions in their models in order to avoid data leakage. Equally important, benchmarking model performance should be carried out between models developed for the same use case. READ MORE

GMP-Featurizer is a lightweight, accurate, efficient, and scalable software package for calculating the Gaussian Multipole (GMP) features (Lei & Medford, 2022) for a variety of atomic systems with elements across the periodic table. Starting from the GMP feature computation module from AmpTorch (AMPTorch, 2020), the capability of GMP-Featurizer has since been greatly improved, including its accuracy and efficiency (please refer to the Overview section for details), as well as the ability to parallelize on different cores, even machines. Moreover, this Python package only has very few dependencies that are all standard Python libraries, plus CFFI for C++ code interfacing and Ray (Moritz et al., 2018) for parallelization, making it lightweight and robust. A set of unit tests are designed to ensure the reliability of its outputs. A set of extensive examples and tutorials, as well as two sets of pseudopotential files (needed for specifying the GMP feature set), are also included in this package for its users. Overall, this package is designed to serve as a standard implementation for chemical and material scientists who are interested in developing models based on GMP features. The source code for this package is freely available to the public under the Apache 2.0 license. READ MORE
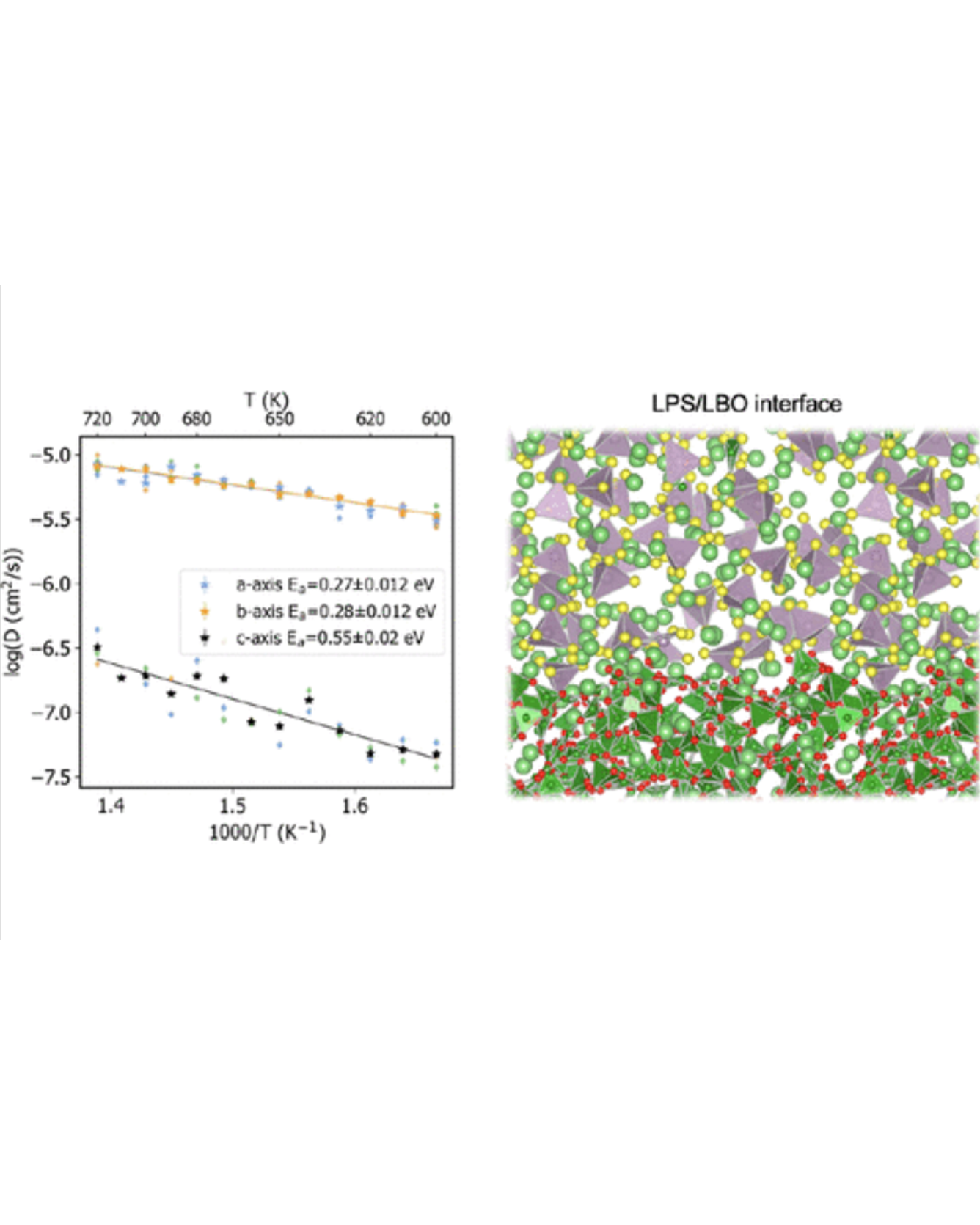
Non-crystalline solid materials have attracted growing attention in energy storage for their desirable properties such as ionic conductivity, stability, and processability. However, compared to bulk crystalline materials, fundamental understanding of these highly complex metastable systems is hindered by the scale limitations of density functional theory (DFT) calculations and resolution limitations of experimental methods. To fill the knowledge gap and guide the rational design of amorphous battery materials and interfaces, we present a molecular dynamics (MD) framework based on machine-learned interatomic potentials trained on the fly to study the amorphous solid electrolyte Li3PS4 and its protective coating, amorphous Li3B11O18. The use of machine-learned potentials allows us to simulate the materials at time and length scales that are not accessible to DFT while maintaining a near-DFT level of accuracy. This approach allows us to calculate amorphization energies, amorphous–amorphous interface energies, and the impact of the interface on lithium ion conductivity. This study demonstrates the promising role of actively learned interatomic potentials in extending the application of ab initio modeling to more complex and realistic systems such as amorphous materials and interfaces. READ MORE
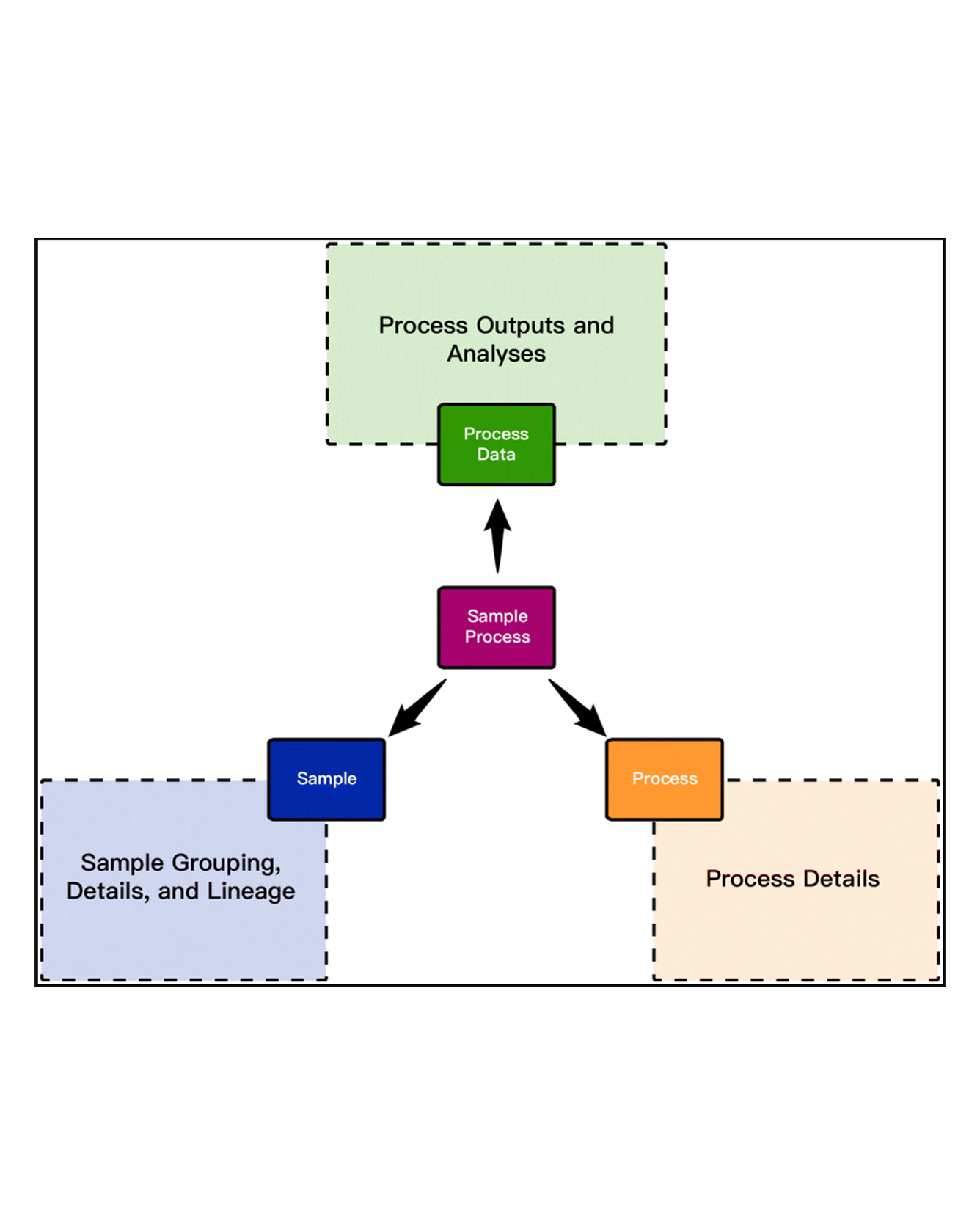
While the vision of accelerating materials discovery using data driven methods is well-founded, practical realization has been throttled due to challenges in data generation, ingestion, and materials state-aware machine learning. High-throughput experiments and automated computational workflows are addressing the challenge of data generation, and capitalizing on these emerging data resources requires ingestion of data into an architecture that captures the complex provenance of experiments and simulations. In this manuscript, we describe an event-sourced architecture for materials provenance (ESAMP) that encodes the sequence and interrelationships among events occurring in a simulation or experiment. We use this architecture to ingest a large and varied dataset (MEAD) that contains raw data and metadata from millions of materials synthesis and characterization experiments performed using various modalities such as serial, parallel, multi-modal experimentation. Our data architecture tracks the evolution of a material's state, enabling a demonstration of how state-equivalency rules can be used to generate datasets that significantly enhance data-driven materials discovery. Specifically, using state-equivalency rules and parameters associated with state-changing processes in addition to the typically used composition data, we demonstrated marked reduction of uncertainty in prediction of overpotential for oxygen evolution reaction (OER) catalysts. Finally, we discuss the importance of ESAMP architecture in enabling several aspects of accelerated materials discovery such as dynamic workflow design, generation of knowledge graphs, and efficient integration of simulation and experiment. READ MORE