Featured Publications

All Publications
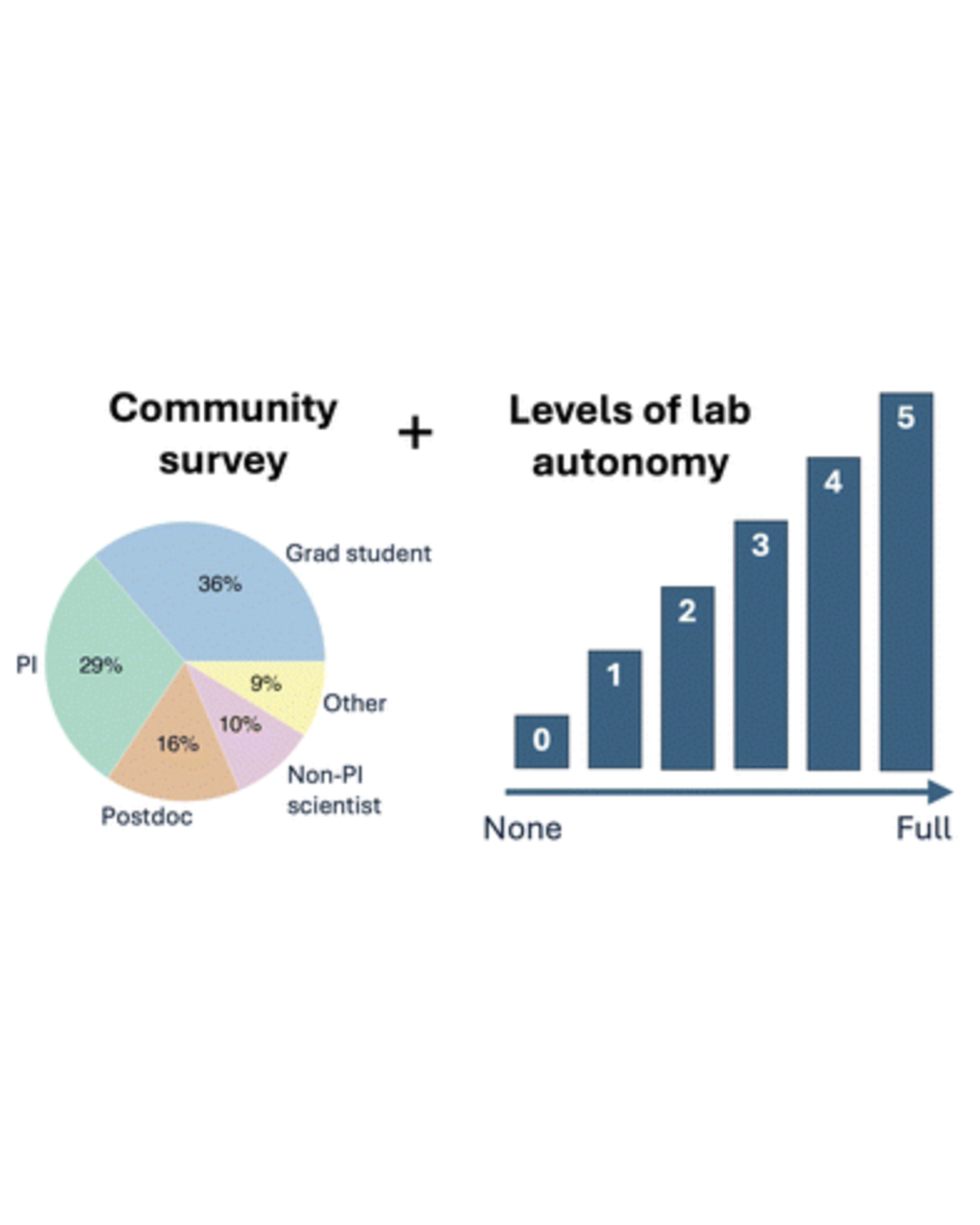
What are researchers' motivations and challenges related to automation and autonomy in materials science laboratories? Our survey on this topic received 102 responses from researchers across a variety of institutions and in a variety of roles. Accelerated discovery was a clear theme in the responses, and another theme was concern about the role of human researchers. Survey respondents shared a variety of use cases targeting accelerated materials discovery, including examples where partial automation is preferred over full self-driving laboratories. Building on the observed patterns of researcher priorities and needs, we propose a framework for levels of laboratory autonomy from non-automated (L0) to fully autonomous (L5). READ MORE
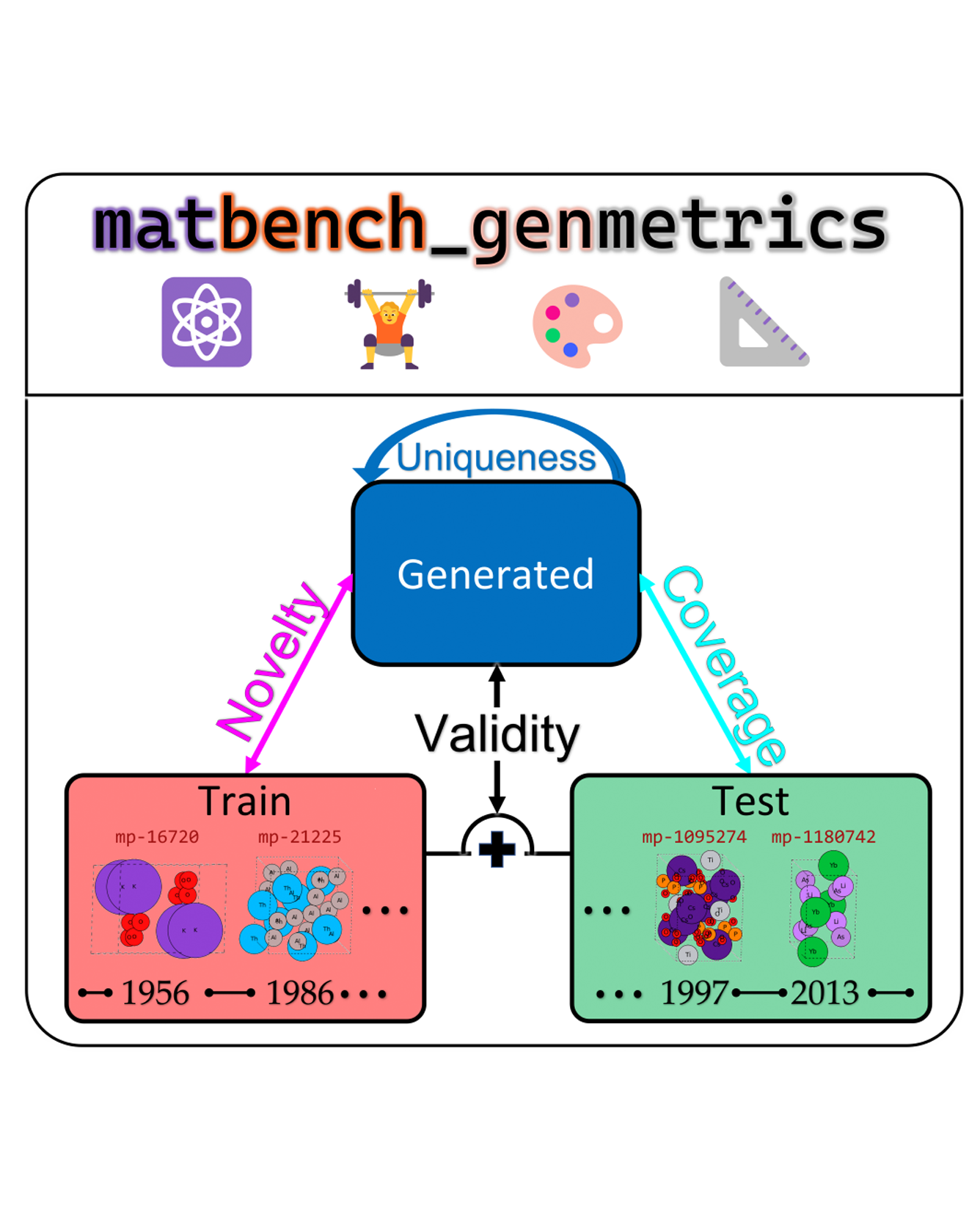
The progress of a machine learning field is both tracked and propelled through the development of robust benchmarks. While significant progress has been made to create standardized, easy-to-use benchmarks for molecular discovery e.g., (Brown et al., 2019), this remains a challenge for solid-state material discovery (Alverson et al., 2024; Xie et al., 2022; Zhao et al., 2023). To address this limitation, we propose matbench-genmetrics, an open-source Python library for benchmarking generative models for crystal structures. We use four evaluation metrics inspired by Guacamol (Brown et al., 2019) and Crystal Diffusion Variational AutoEncoder (CDVAE) (Xie et al., 2022)—validity, coverage, novelty, and uniqueness—to assess performance on Materials Project data splits using timeline-based cross-validation. We believe that matbench-genmetrics will provide the standardization and convenience required for rigorous benchmarking of crystal structure generative models. READ MORE
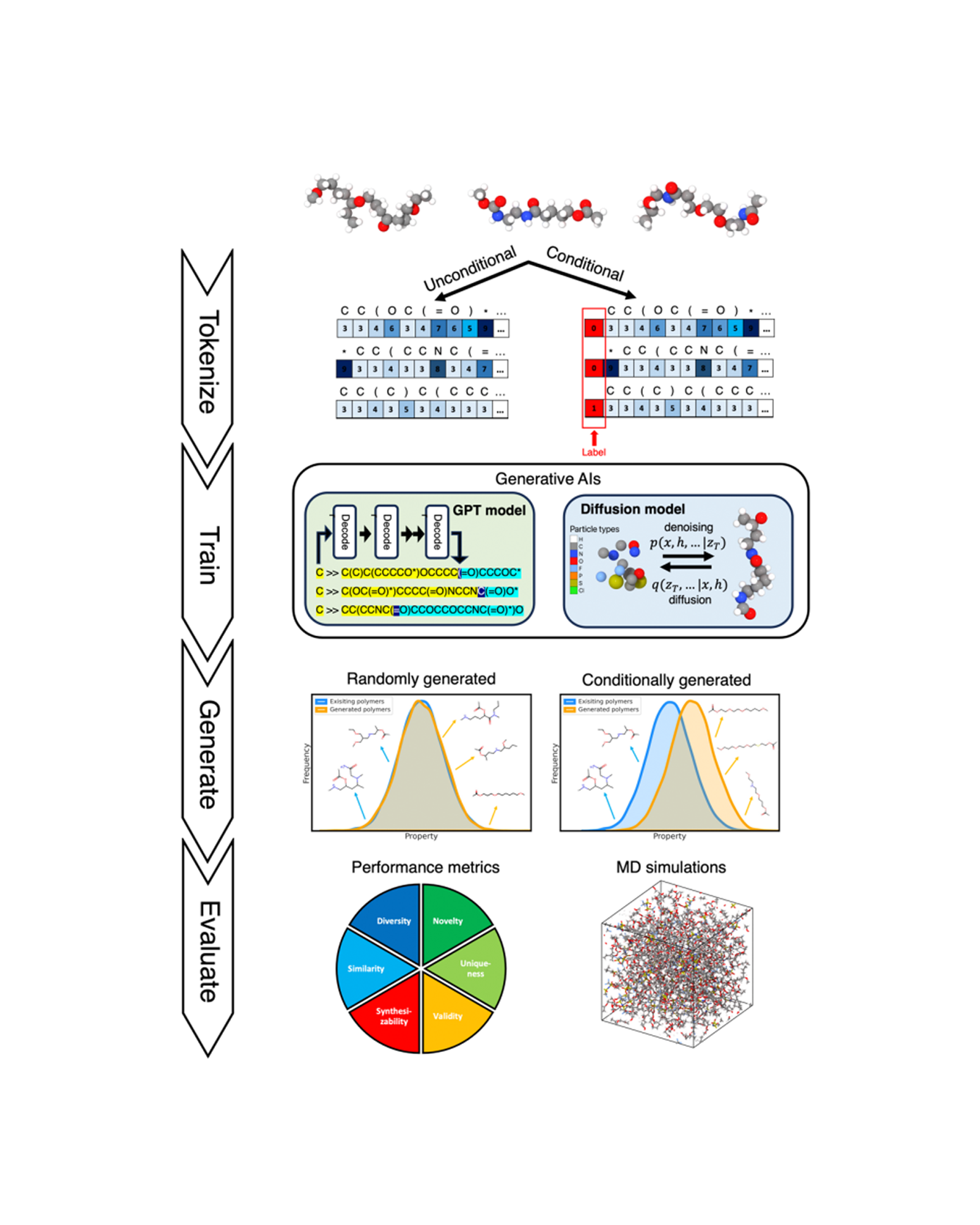
Solid polymer electrolytes hold significant promise as materials for next-generation batteries due to their superior safety performance, enhanced specific energy, and extended lifespans compared to liquid electrolytes. However, the material's low ionic conductivity impedes its commercialization, and the vast polymer space poses significant challenges for the screening and design. In this study, we assess the capabilities of generative artificial intelligence (AI) for the de novo design of polymer electrolytes. To optimize the generation, we compare different deep learning architectures, including both GPT-based and diffusion-based models, and benchmark the results with hyperparameter tuning. We further employ various evaluation metrics and full-atom molecular dynamics simulations to assess the performance of different generative model architectures and to validate the top candidates produced by each model. Out of only 45 candidates being tested, we discovered 17 polymers that achieve superior ionic conductivity better than any other polymers in our database, with some of them doubling the conductivity value. In addition, by adopting a pretraining and fine-tuning methodology, we significantly improve the efficacy of our generative models, achieving quicker convergence, enhanced performance with limited data, and greater diversity. Using the proposed method, we can easily generate a large number of novel, diverse, and valid polymers, with a chance of synthesizability, enabling us to identify promising candidates with markedly improved efficiency. READ MORE
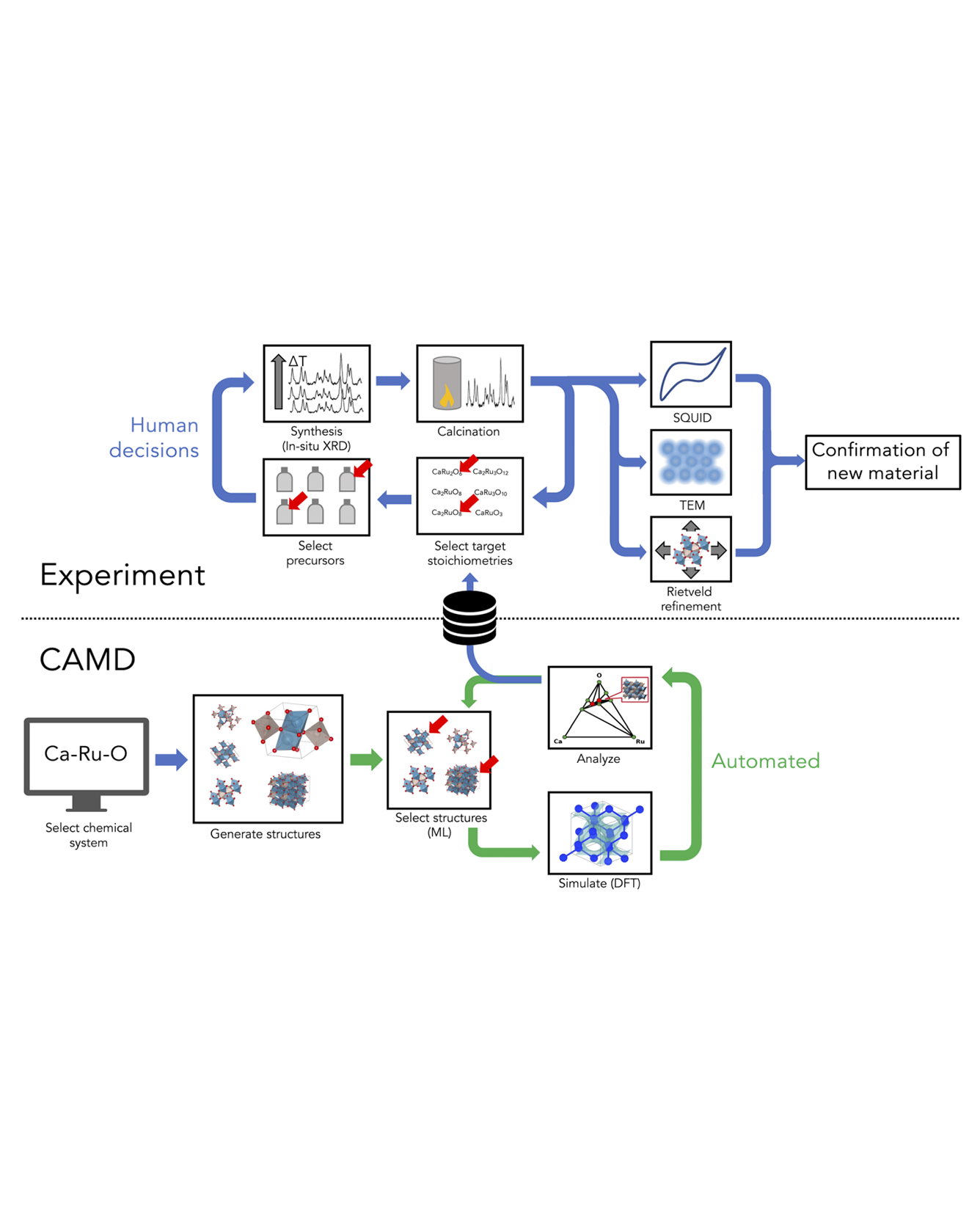
Exploratory synthesis has been the main generator of new inorganic materials for decades. However, our Edisonian and bias-prone processes of synthetic exploration alone are no longer sufficient in an age that demands rapid advances in materials development. In this work, we demonstrate an end-to-end attempt towards systematic, computer-aided discovery and laboratory synthesis of inorganic crystalline compounds as a modern alternative to purely exploratory synthesis. Our approach initializes materials discovery campaigns by autonomously mapping the synthetic feasibility of a chemical system using density functional theory with AI feedback. Following expert-driven down-selection of newly generated phases, we use solid-state synthesis and in situ characterization via hot-stage X-ray diffraction in order to realize new ternary oxide phases experimentally. We applied this strategy in six ternary transition-metal oxide chemistries previously considered well-explored, one of which culminated in the discovery of two novel phases of calcium ruthenates. Detailed characterization using room temperature X-ray powder diffraction, 4D-STEM and SQUID measurements identifies the structure and composition and confirms distinct properties, including distinct defect concentrations, of one of the new phases formed in our experimental campaigns. While the discovery of a new material guided by AI and DFT theory represents a milestone, our procedure and results also highlight a number of critical gaps in the process that can inform future efforts towards the improvement of AI-coupled methodologies. READ MORE
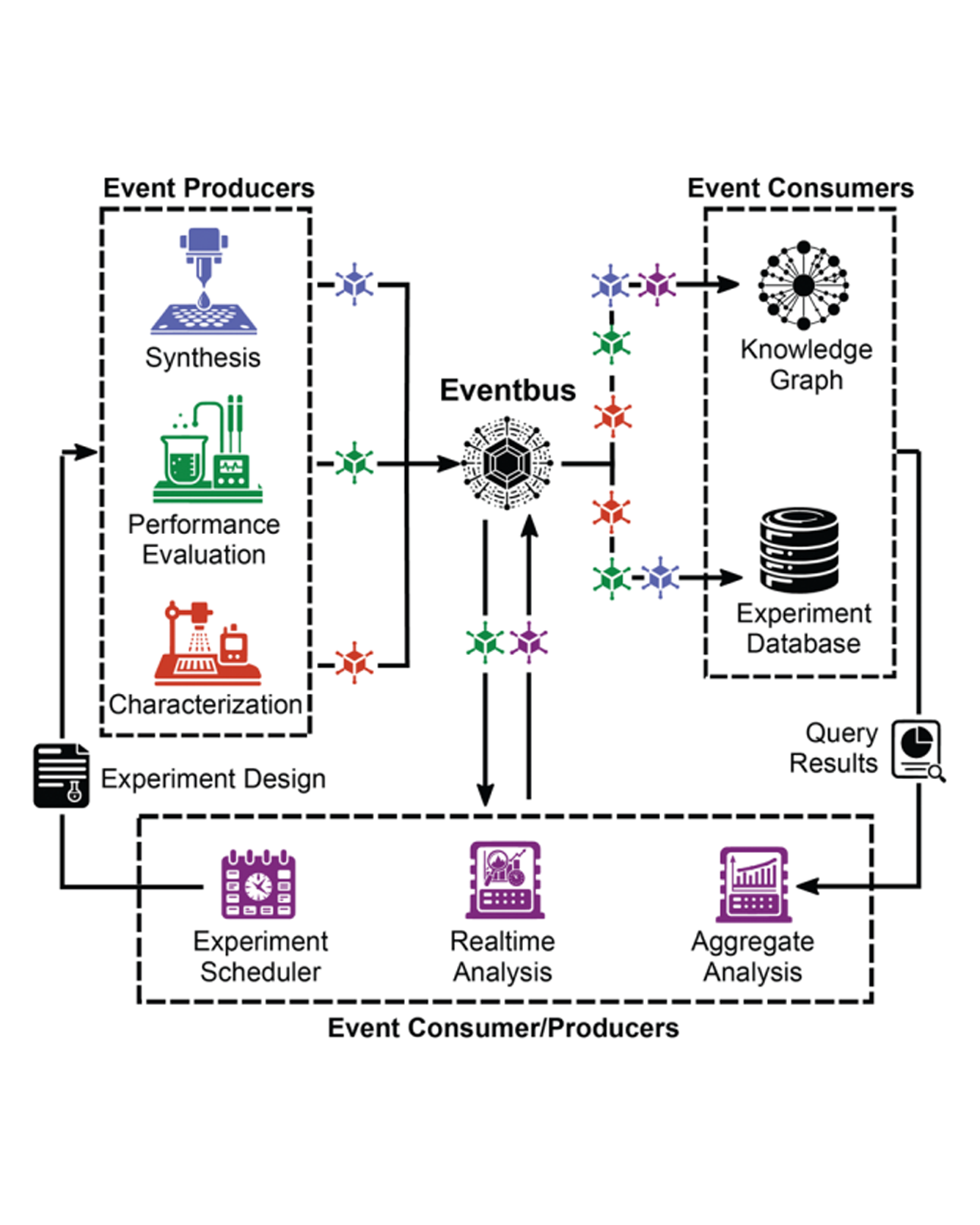
The materials research community is increasingly using automation and artificial intelligence (AI) to accelerate research and development. A materials acceleration platform (MAP) typically encompasses several experimental techniques or instruments to establish a synthesis-characterization-evaluation workflow. With the advancement of workflow orchestration software and AI experiment design, the scope and complexity of MAPs are increasing, however each MAP typically operates as a standalone entity with dedicated experiment, compute, and database resources. The data from each MAP is thus siloed until subsequent efforts to integrate data into complex schema such as knowledge graphs. To lower the latency of data integration and establish an extensible community of MAPs, we must expand our automation efforts to include data handling that is decoupled from the resources of each MAP. Event-driven pipelines are well established in the computational community for building decoupled data processing systems. Such pipelines can be difficult to implement de novo due to their distributed nature and complex error handling. Fortunately, the broader computational science community has established a suite of cloud services that are well suited for this task. By leveraging cloud computing resources to establish event-driven data management, the MAP community can better realize the ideals of extensibility and interoperability in materials chemistry research. READ MORE
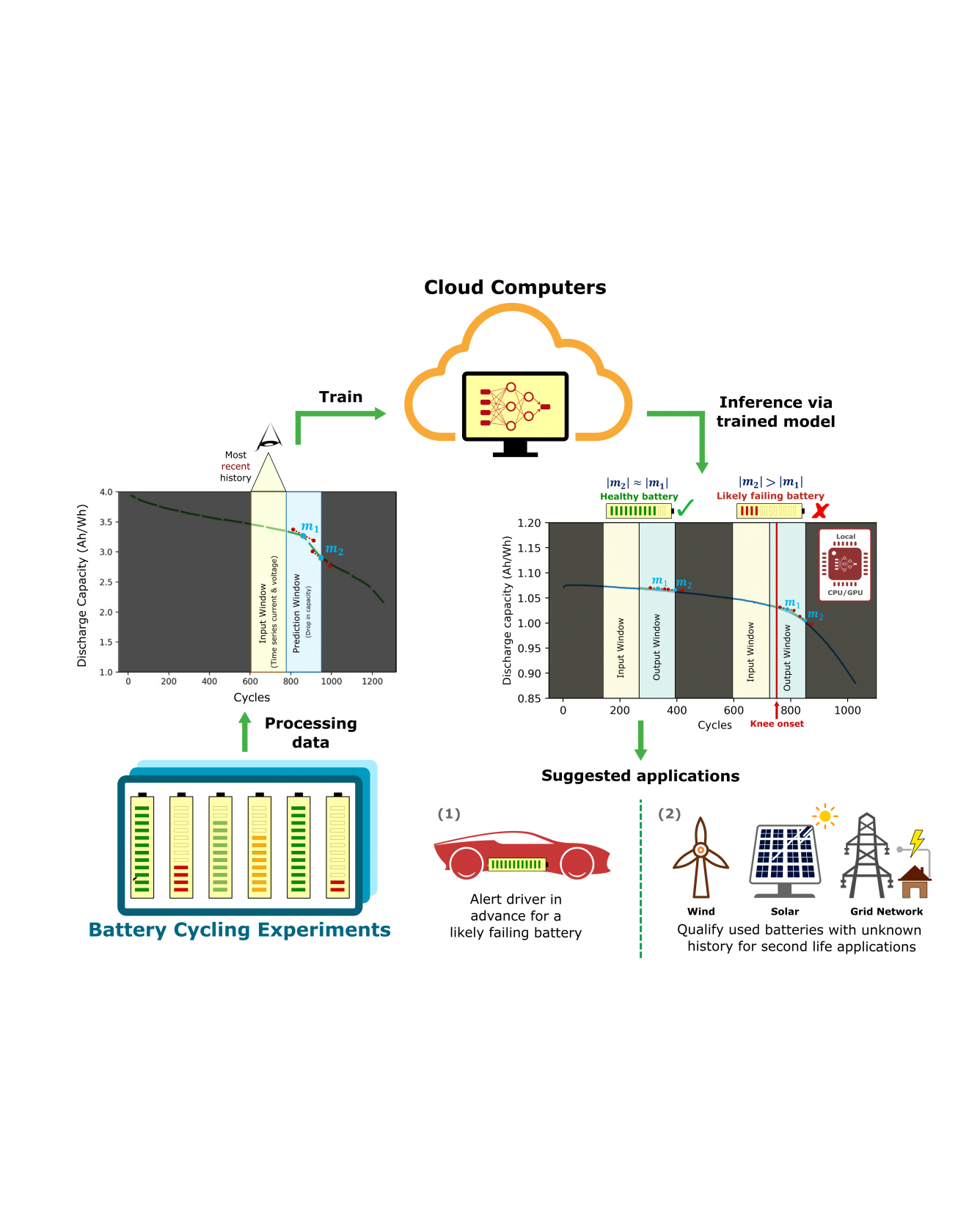
Lithium-ion batteries (LIBs) have attracted widespread attention as an efficient energy storage device on electric vehicles (EV) to achieve emission-free mobility. However, the performance of LIBs deteriorates with time and usage, and the state of health of used batteries are difficult to quantify. Having accurate estimations of a battery’s remaining life across different life stages would benefit maintenance, safety, and serve as a means of qualifying used batteries for second-life applications. Since the full history of a battery may not always be available in downstream applications, in this study, we demonstrate a deep learning framework that enables dynamic degradation rate prediction, including both short-term and long-term forecasting, while requiring only the most recent battery usage information. Specifically, our model takes a rolling window of current and voltage time-series inputs, and predicts the near-term and long-term capacity fade via a recurrent neural network. We exhaustively benchmark our model against a naive extrapolating model by evaluating the error on reconstructing the discharge capacity profile under different settings. We show that our model’s performance in accurately inferring the battery’s degradation profile is agnostic with respect to cell cycling history and its current state of health. This approach can provide a promising path towards evaluating battery health in running vehicles, enhance edge-computing battery diagnostics, and determine the state of health for used batteries with unknown cycling histories. READ MORE
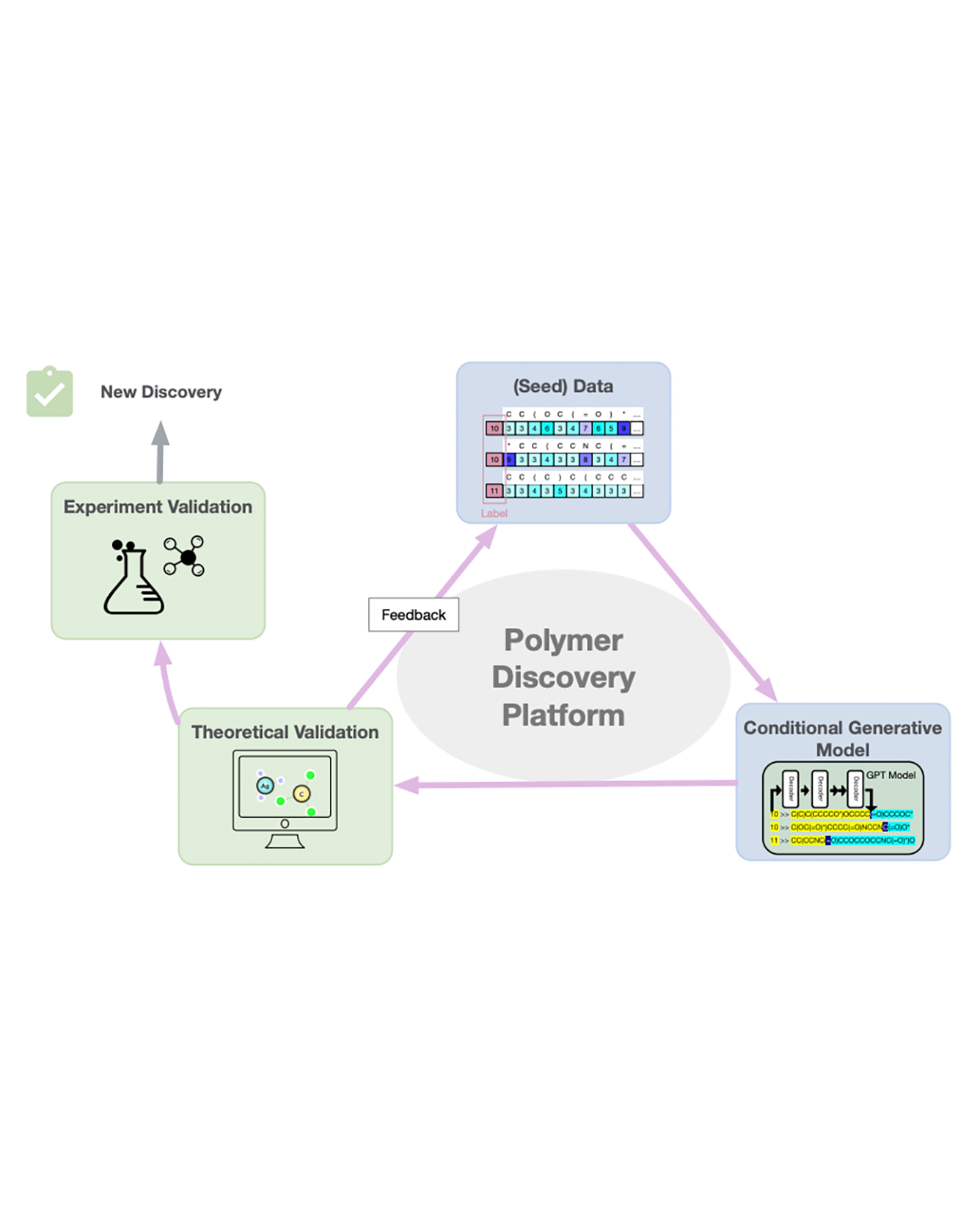
In this work, we introduce a polymer discovery platform designed to identify polymers with tailored properties efficiently, exemplified through the discovery of high-performance polymer electrolytes. The platform integrates three core components: a conditioned generative model, validation modules, and a feedback mechanism, creating a self-improving system for material innovation. To demonstrate the efficacy of this platform, it is used to identify polymer electrolyte materials with high ionic conductivity. A simple conditional generative model, based on the minGPT architecture, can effectively generate candidate polymers that exhibit a mean ionic conductivity that is significantly greater than those in the original training set. This approach, coupled with molecular dynamics simulations for validation and a specifically designed acquisition mechanism, allows the platform to refine its output iteratively. Notably, after the first iteration, we observed an increase in both the mean and the lower bound of the ionic conductivity of the new polymer candidates. The platform's effectiveness is underscored by the identification of 19 polymer repeating units, each displaying a computed ionic conductivity surpassing that of Polyethylene Oxide (PEO). The discovery of these polymers validates the platform's efficacy in identifying potential polymer materials. Acknowledging current limitations, future work will focus on enhancing modeling techniques, validation processes, and acquisition strategies, aiming for broader applicability in polymer science and machine learning. READ MORE
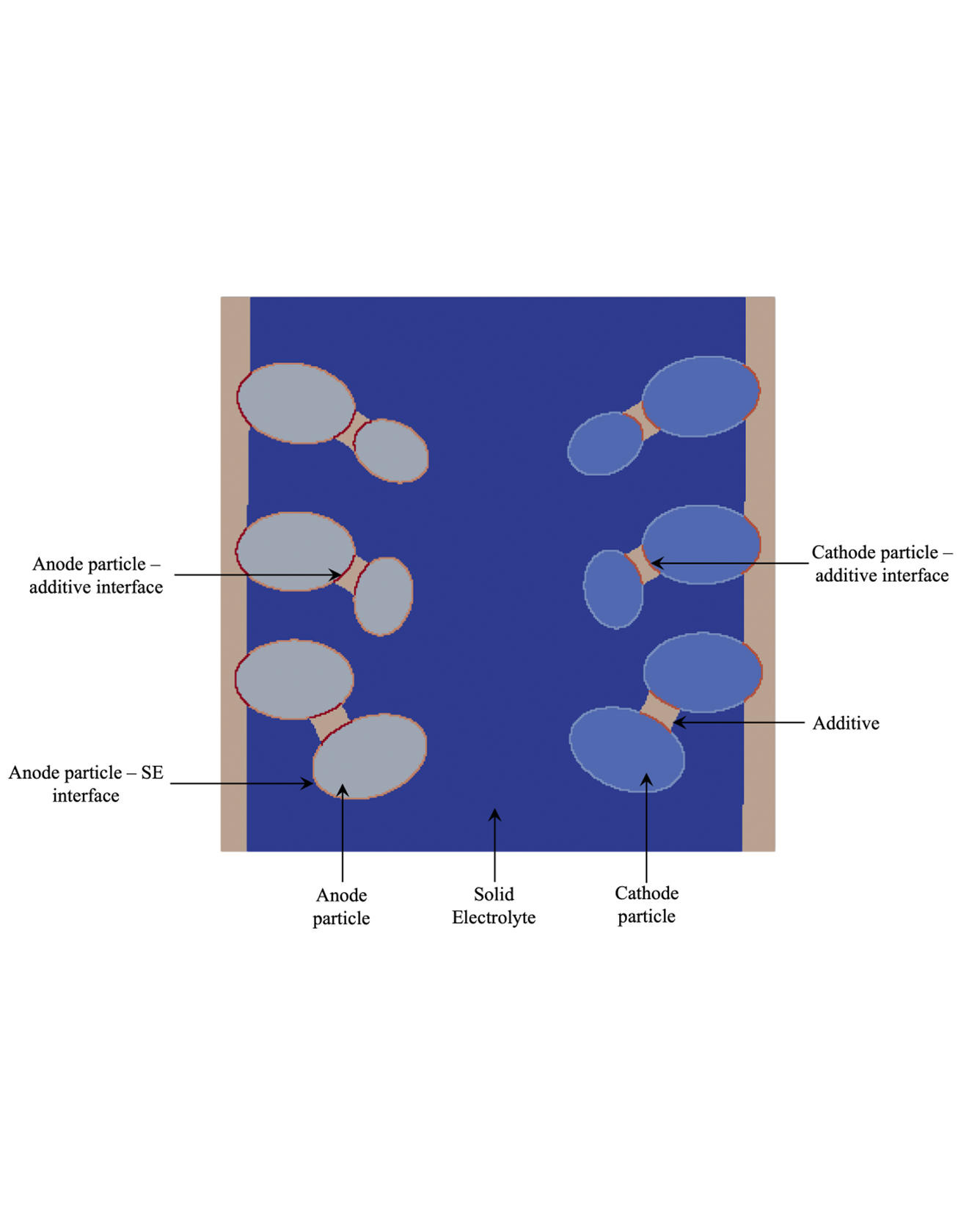
In this work, we present a computational framework for coupled electro-chemo-(nonlinear) mechanics at the particle scale for solid-state batteries. The framework accounts for interfacial fracture between the active particles and solid electrolyte due to intercalation stresses. We extend discontinuous finite element methods for a sharp interface treatment of discontinuities in concentrations, fluxes, electric fields and in displacements, the latter arising from active particle–solid electrolyte interface fracture. We model the degradation in the charge transfer process that results from the loss of contact due to fracture at the electrolyte–active particle interfaces. Additionally, we account for the stress-dependent kinetics that can influence the charge transfer reactions and solid state diffusion. The discontinuous finite element approach does not require a conformal mesh. This offers the flexibility to construct arbitrary particle shapes and geometries that are based on design, or are obtained from microscopy images. The finite element mesh, however, can remain Cartesian, and independent of the particle goemetries. We demonstrate this computational framework on micro-structures that are representative of solid-state batteries with single and multiple anode and cathode particles. READ MORE
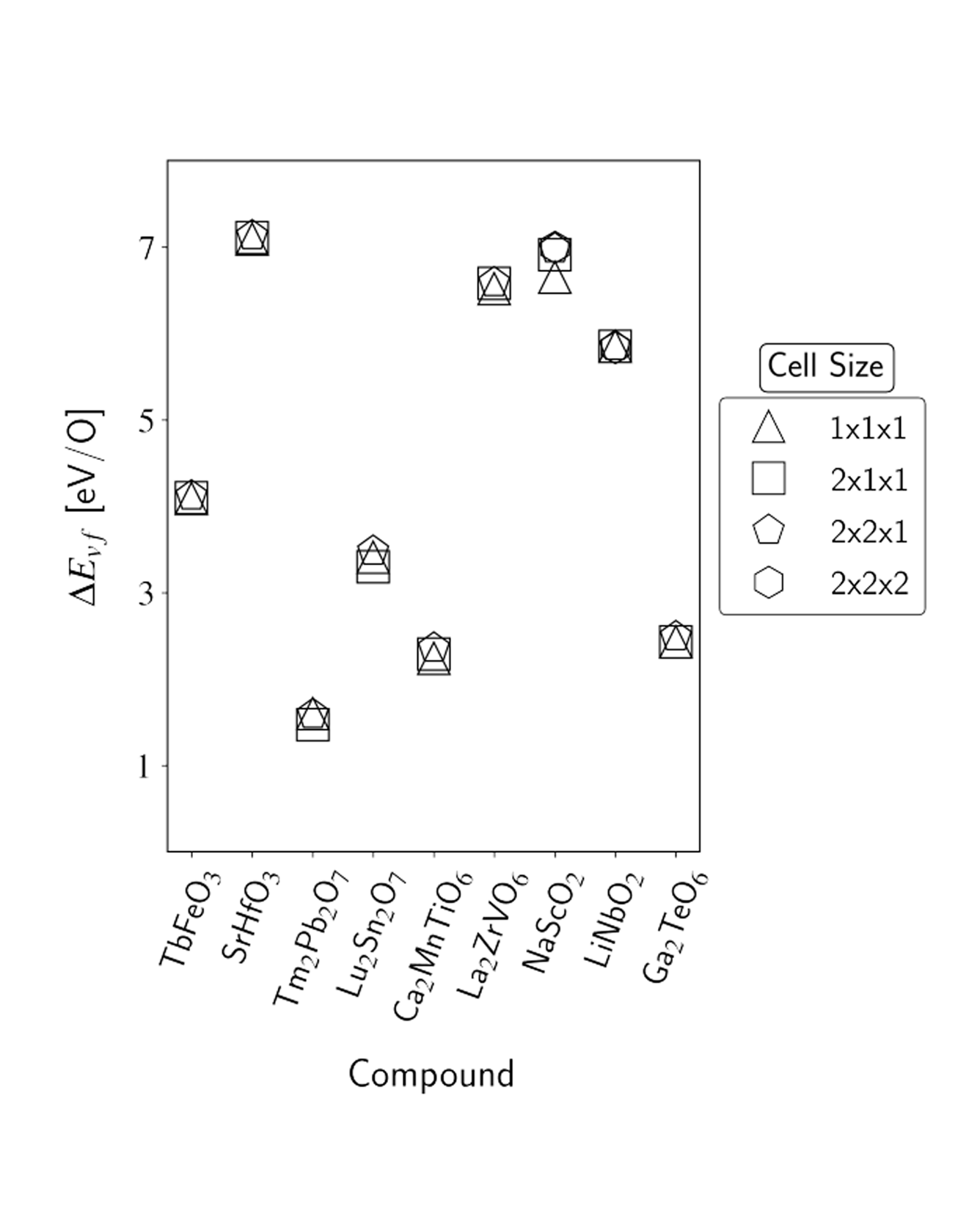
The oxygen vacancy formation energy (ΔEvf) governs defect concentrations alongside the entropy and is a useful metric to perform materials selection for a variety of applications. However, density functional theory (DFT) calculations of ΔEvf come at a greater computational cost than the typical bulk calculations available in materials databases due to the involvement of multiple vacancy-containing supercells. As a result, available repositories of direct calculations of ΔEvf remain relatively scarce, and the development of machine-learning models capable of delivering accurate predictions is of interest. In the present work, we address both such points. We first report the results of new high-throughput DFT calculations of oxygen vacancy formation energies of the different unique oxygen sites in over 1000 different oxide materials, with a large portion of the calculations, and of the discussion, focusing on perovskite-type and pyrochlore-type oxides. Together, the over 2500 ΔEvf calculations form the largest data set of directly computed oxygen vacancy formation energies to date, to our knowledge. We then utilize such a data set to train random forest models with different sets of features, examining both novel features introduced in this work and ones previously employed in the literature. We demonstrate the benefits of including features that contain information specific to the vacancy site and account for both cation identity and oxidation state and achieve a mean absolute error upon prediction of ∼0.3 eV/O, which is comparable to the accuracy observed upon comparison of DFT computations of oxygen vacancy formation energy and experimental results. Finally, we exemplify the predictive power of the developed models in the search for new compounds for solar-thermochemical water-splitting applications, finding over 250 new AA′BB′O6 double perovskite candidates. READ MORE
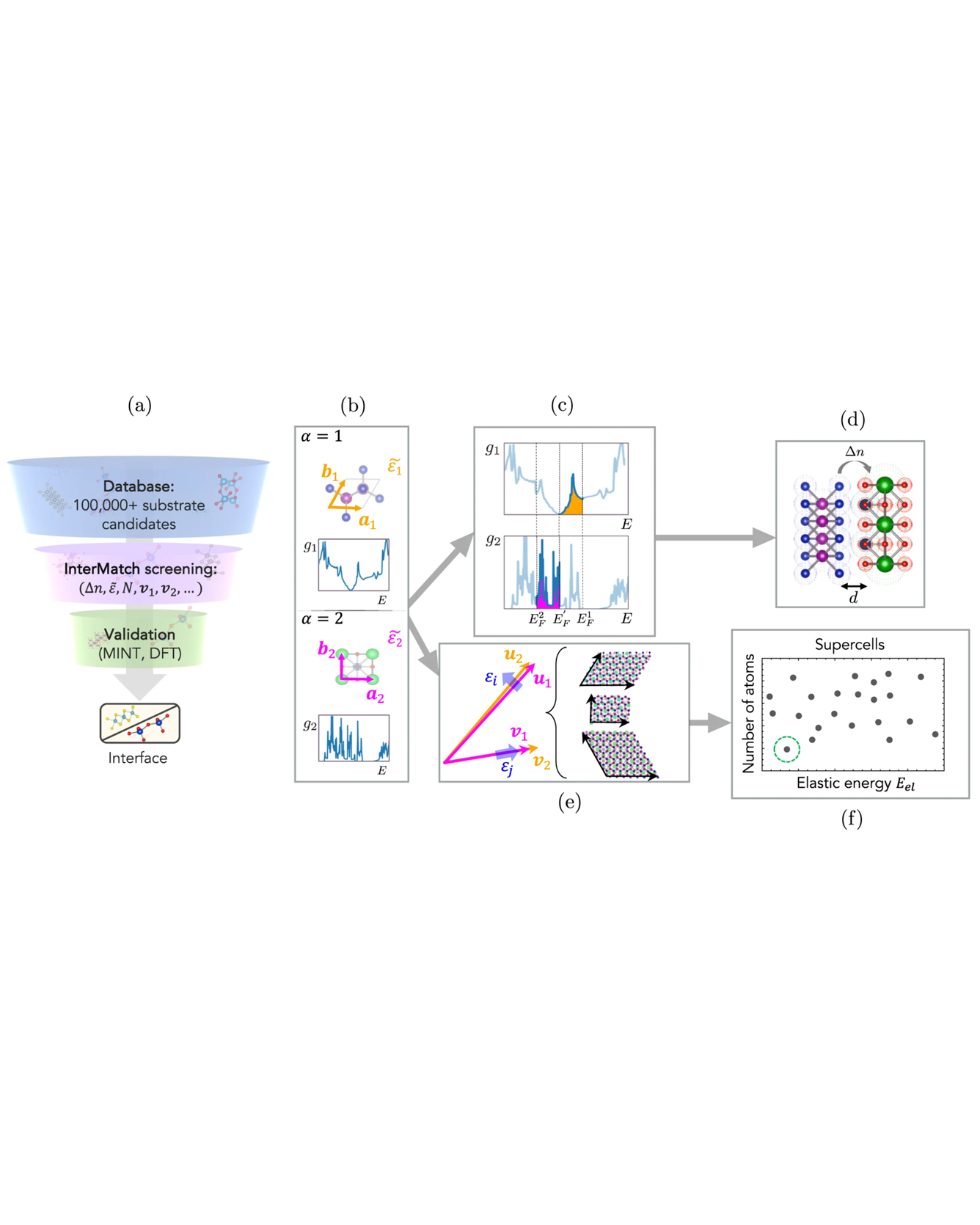
Forming a hetero-interface is a materials-design strategy that can access an astronomically large phase space. However, the immense phase space necessitates a high-throughput approach for an optimal interface design. Here we introduce a high-throughput computational framework, InterMatch, for efficiently predicting charge transfer, strain, and superlattice structure of an interface by leveraging the databases of individual bulk materials. Specifically, the algorithm reads in the lattice vectors, density of states, and the stiffness tensors for each material in their isolated form from the Materials Project. From these bulk properties, InterMatch estimates the interfacial properties. We benchmark InterMatch predictions for the charge transfer against experimental measurements and supercell density-functional theory calculations. We then use InterMatch to predict promising interface candidates for doping transition metal dichalcogenide MoSe2. Finally, we explain experimental observation of factor of 10 variation in the supercell periodicity within a few microns in graphene/α-RuCl3 by exploring low energy superlattice structures as a function of twist angle using InterMatch. We anticipate our open-source InterMatch algorithm accelerating and guiding ever-growing interfacial design efforts. Moreover, the interface database resulting from the InterMatch searches presented in this paper can be readily accessed online. READ MORE