
Featured Publications

All Publications
The discovery of high-performing and stable materials for sustainable energy applications is a pressing goal in catalysis and materials science. Understanding the relationship between a material’s structure and functionality is an important step in the process, such that viable polymorphs for a given chemical composition need to be identified. Machine-learning-based surrogate models have the potential to accelerate the search for polymorphs that target specific applications. Herein, we report a readily generalizable active-learning (AL) accelerated algorithm for identification of electrochemically stable iridium oxide polymorphs of IrO2 and IrO3. The search is coupled to a subsequent analysis of the electrochemical stability of the discovered structures for the acidic oxygen evolution reaction (OER). Structural candidates are generated by identifying all 956 structurally unique AB2 and AB3 prototypes in existing materials databases (more than 38000). Next, using an active learning approach, we find 196 IrO2 polymorphs within the thermodynamic amorphous synthesizability limit and reaffirm the global stability of the rutile structure. We find 75 synthesizable IrO3 polymorphs and report a previously unknown FeF3-type structure as the most stable, termed α-IrO3. To test the algorithms performance, we compare to a random search of the candidate space and report at least a 2-fold increase in the rate of discovery. Additionally, the AL approach can acquire the most stable polymorphs of IrO2 and IrO3 with fewer than 30 density functional theory optimizations. Analysis of the structural properties of the discovered polymorphs reveals that octahedral local coordination environments are preferred for nearly all low-energy structures. Subsequent Pourbaix Ir–H2O analysis shows that α-IrO3 is the globally stable solid phase under acidic OER conditions and supersedes the stability of rutile IrO2. Calculation of theoretical OER surface activities reveal ideal weaker binding of the OER intermediates on α-IrO3 than on any other considered iridium oxide. We emphasize that the proposed AL algorithm can be easily generalized to search for any binary metal oxide structure with a defined stoichiometry. READ MORE
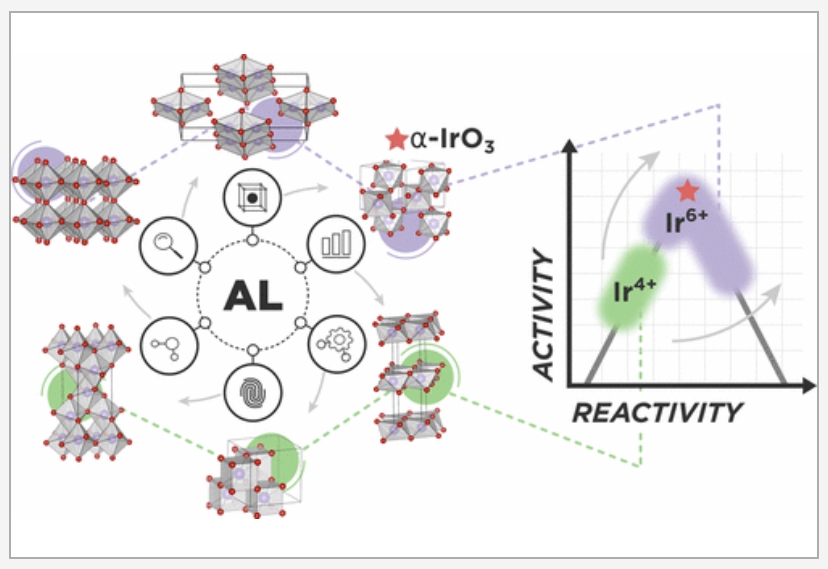
TRI Authors: Muratahan Aykol, Patrick Herring, & Abraham Anapolsky All Authors: Muratahan Aykol, Patrick Herring, & Abraham Anapolsky
Batteries, as complex materials systems, pose unique challenges for the application of machine learning. Although a shift to data-driven, machine learning-based battery research has started, new initiatives in academia and industry are needed to fully exploit its potential. Read more
Citation: Aykol, Muratahan, Patrick Herring, Abraham Anapolsky. “Machine learning for continuous innovation in battery technologies.” Nature Reviews Materials (2020). https://doi.org/10.1038/s41578-020-0216-y
Hydrogen peroxide is a valuable chemical oxidant with a wide range of applications in a variety of industrial processes, especially in water sanitization. Electrochemical synthesis of hydrogen peroxide (H2O2) through a two-electron oxygen reduction reaction (2e-ORR) or a two-electron water oxidation reaction (2e-WOR) has emerged as an appealing process for onsite production of this chemically valuable oxidant. On-site produced H2O2 can be applied for wastewater treatment in remote locations or any applications where H2O2 is needed as an oxidizing agent. This Review studies the theoretical efforts in understanding the challenges in catalysis for electrochemical synthesis of H2O2 as well as providing design principles for more efficient catalyst materials. READ MORE
TRI Authors: Muratahan Aykol, Joseph Montoya, Jens Hummelshøj
All Authors: Roni Choudhury, Muratahan Aykol, Samuel Gratzl, Joseph Montoya, Jens Hummelshøj
Materials science research deals primarily with understanding the relationship between the structure and properties of materials. With recent advances in computational power and automation of simulation techniques, material structure and property databases have emerged (Curtarolo et al., 2012; Jain et al., 2013; Kirklin et al., 2015), allowing a more data-driven approach to carrying out materials research. Recent studies have demonstrated that representing these databases as material networks can enable extraction of new materials knowledge (Hegde, Aykol, Kirklin, & Wolverton, 2018; Isayev et al., 2015) or help tackle challenges like predictive synthesis (Aykol, Hegde, et al., 2019) that require relational information between materials. Materials databases have become very popular because they enable their users to do rapid prototyping by searching near globally for figures of merit for their target application. However, both scientists and engineers have little in the way of visualization of aggregates from these databases, that is, intuitive layouts that help understand which materials are related and how they are related. The need for a tool that does this is particularly crucial in materials science because properties like phase stability and crystal structure similarity are themselves functions of a material dataset, rather than of individual materials. Read More
Citation: Choudhury, Roni, Muratahan Aykol, Samuel Gratzl, Joseph Montoya, and Jens Hummelshøj. "MaterialNet: A web-based graph explorer for materials science data." Journal of Open Source Software 5, no. 47 (2020): 2105.

TRI Authors: Muratahan Aykol*
All Authors: Vinay Hegde, Muratahan Aykol*, Scott Kirklin, Chris Wolverton*
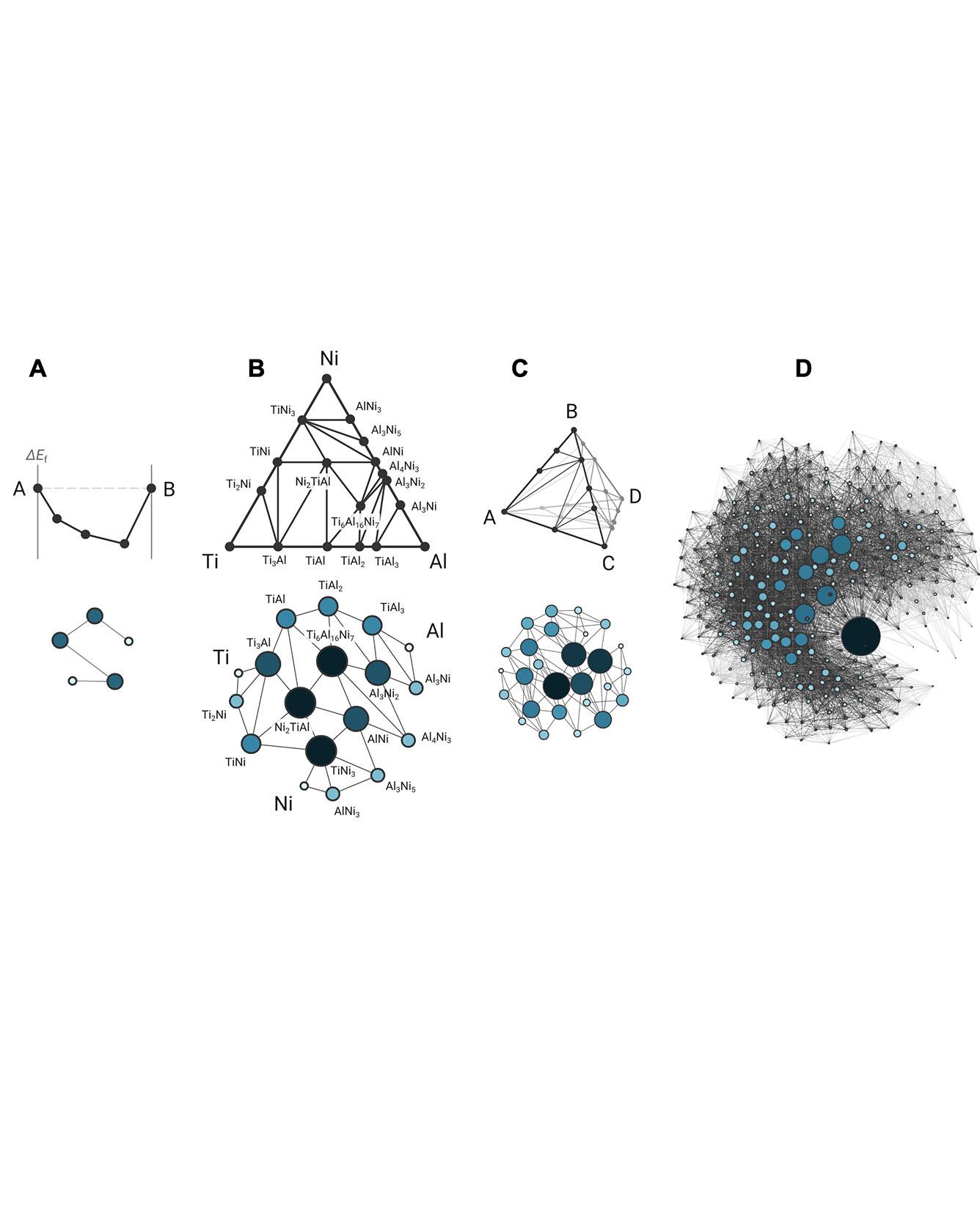
One of the holy grails of materials science, unlocking structure-property relationships, has largely been pursued via bottom-up investigations of how the arrangement of atoms and interatomic bonding in a material determine its macroscopic behavior. Here, we consider a complementary approach, a top-down study of the organizational structure of networks of materials, based on the interaction between materials themselves. We unravel the complete “phase stability network of all inorganic materials” as a densely connected complex network of 21,000 thermodynamically stable compounds (nodes) interlinked by 41 million tie line (edges) defining their two-phase equilibria, as computed by high-throughput density functional theory. Analyzing the topology of this network of materials has the potential to uncover previously unidentified characteristics inaccessible from traditional atoms-to-materials paradigms. Using the connectivity of nodes in the phase stability network, we derive a rational, data-driven metric for material reactivity, the “nobility index,” and quantitatively identify the noblest materials in nature. Read More
Citation: Hegde, Vinay I., Muratahan Aykol, Scott Kirklin, and Chris Wolverton. "The phase stability network of all inorganic materials." Science Advances 6, no. 9 (2020): eaay5606.
TRI Authors: Patrick K Herring, Muratahan Aykol
All Authors: Peter M Attia, Aditya Grover, Norman Jin, Kristen A Severson, Todor M Markov, Yang-Hung Liao, Michael H Chen, Bryan Cheong, Nicholas Perkins, Zi Yang, Patrick K Herring, Muratahan Aykol, Stephen J Harris, Richard D Braatz, Stefano Ermon, William C Chueh
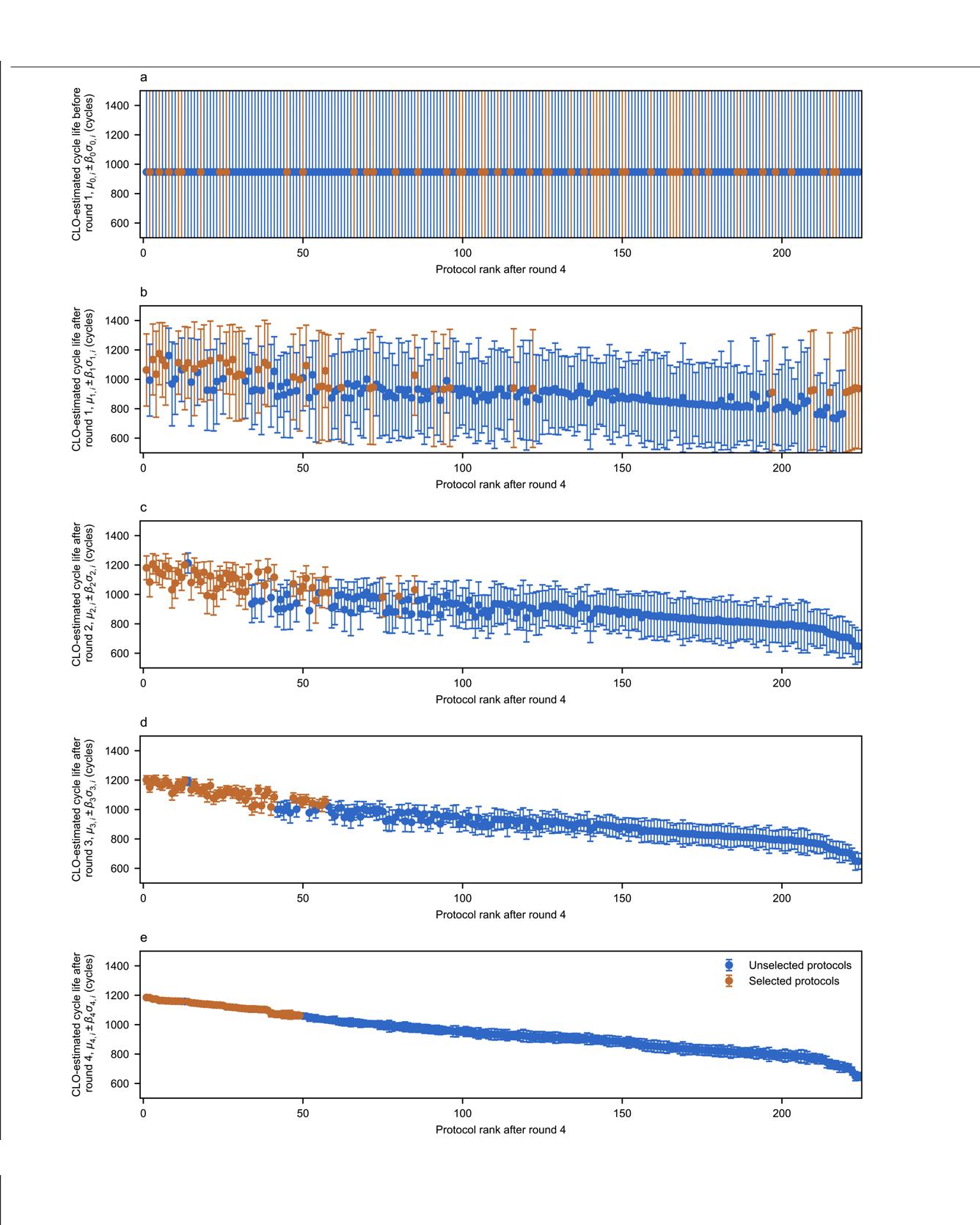
Simultaneously optimizing many design parameters in time-consuming experiments causes bottlenecks in a broad range of scientific and engineering disciplines1,2. One such example is process and control optimization for lithium-ion batteries during materials selection, cell manufacturing and operation. A typical objective is to maximize battery lifetime; however, conducting even a single experiment to evaluate lifetime can take months to years3,4,5. Furthermore, both large parameter spaces and high sampling variability3,6,7 necessitate a large number of experiments. Hence, the key challenge is to reduce both the number and the duration of the experiments required. Here we develop and demonstrate a machine learning methodology to efficiently optimize a parameter space specifying the current and voltage profiles of six-step, ten-minute fast-charging protocols for maximizing battery cycle life, which can alleviate range anxiety for electric-vehicle users8,9. We combine two key elements to reduce the optimization cost: an early-prediction model5, which reduces the time per experiment by predicting the final cycle life using data from the first few cycles, and a Bayesian optimization algorithm10,11, which reduces the number of experiments by balancing exploration and exploitation to efficiently probe the parameter space of charging protocols. Using this methodology, we rapidly identify high-cycle-life charging protocols among 224 candidates in 16 days (compared with over 500 days using exhaustive search without early prediction), and subsequently validate the accuracy and efficiency of our optimization approach. Our closed-loop methodology automatically incorporates feedback from past experiments to inform future decisions and can be generalized to other applications in battery design and, more broadly, other scientific domains that involve time-intensive experiments and multi-dimensional design spaces. Read More
Citation: Attia, Peter M., Aditya Grover, Norman Jin, Kristen A. Severson, Todor M. Markov, Yang-Hung Liao, Michael H. Chen et al. "Closed-loop optimization of fast-charging protocols for batteries with machine learning." Nature 578, no. 7795 (2020): 397-402.
TRI Author: Brian D. Storey
All Authors: Hongbo Zhao, Brian D. Storey, Richard D. Braatz, and Martin Z. Bazant
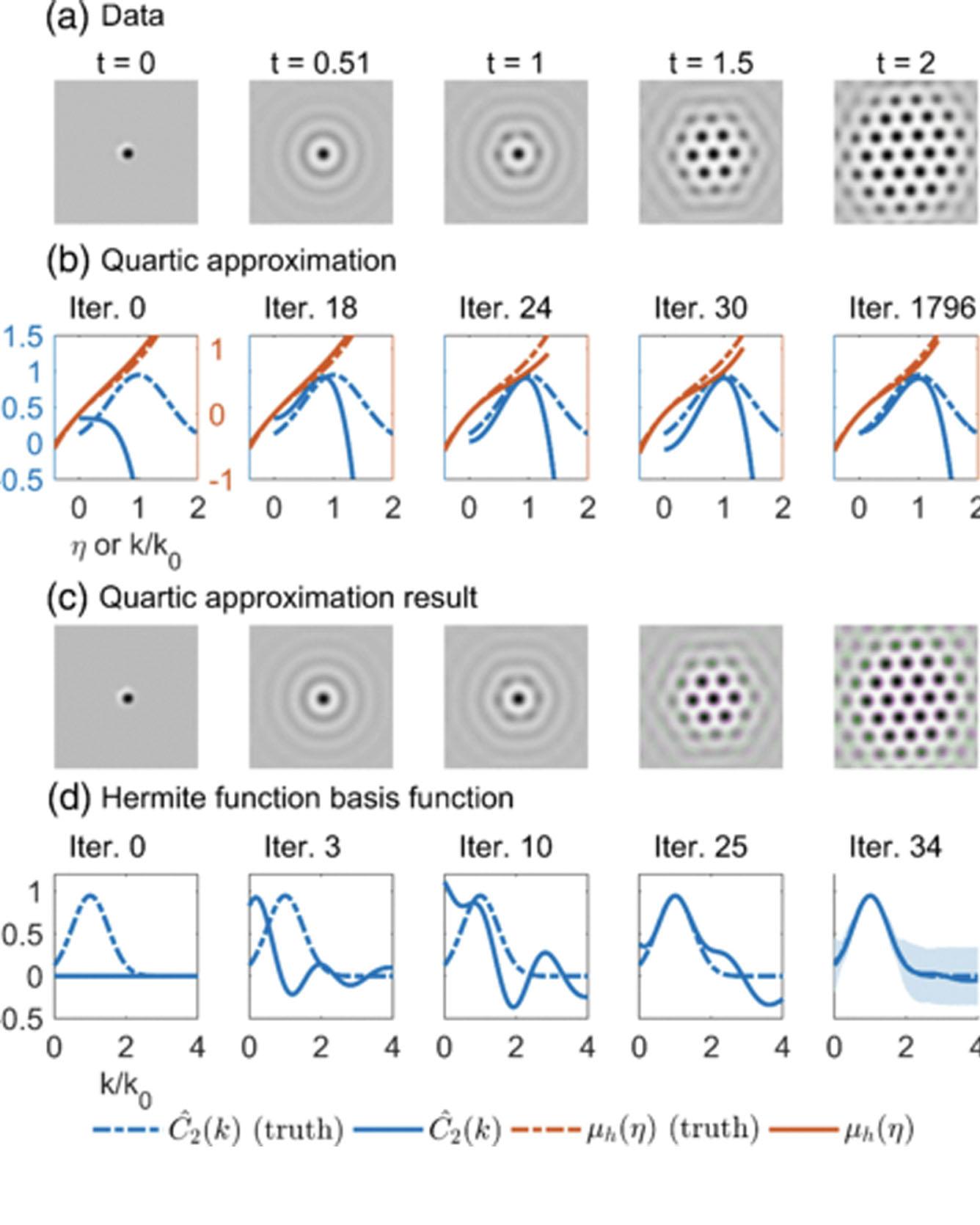
Using a framework of partial differential equation-constrained optimization, we demonstrate that multiple constitutive relations can be extracted simultaneously from a small set of images of pattern formation. Examples include state-dependent properties in phase-field models, such as the diffusivity, kinetic prefactor, free energy, and direct correlation function, given only the general form of the Cahn-Hilliard equation, Allen-Cahn equation, or dynamical density functional theory (phase-field crystal model). Constraints can be added based on physical arguments to accelerate convergence and avoid spurious results. Reconstruction of the free energy functional, which contains nonlinear dependence on the state variable and differential or convolutional operators, opens the possibility of learning nonequilibrium thermodynamics from only a few snapshots of the dynamics. Read More
Citation: Zhao, Hongbo, Brian D. Storey, Richard D. Braatz, and Martin Z. Bazant. "Learning the Physics of Pattern Formation from Images." Physical Review Letters 124, no. 6 (2020): 060201.
TRI Author: Montoya, J. H.
All Authors: Mrdjenovich, D., Horton, M. K., Montoya, J. H., Legaspi, C. M., Dwaraknath, S., Tshitoyan, V., Jain A., Persson, K. A
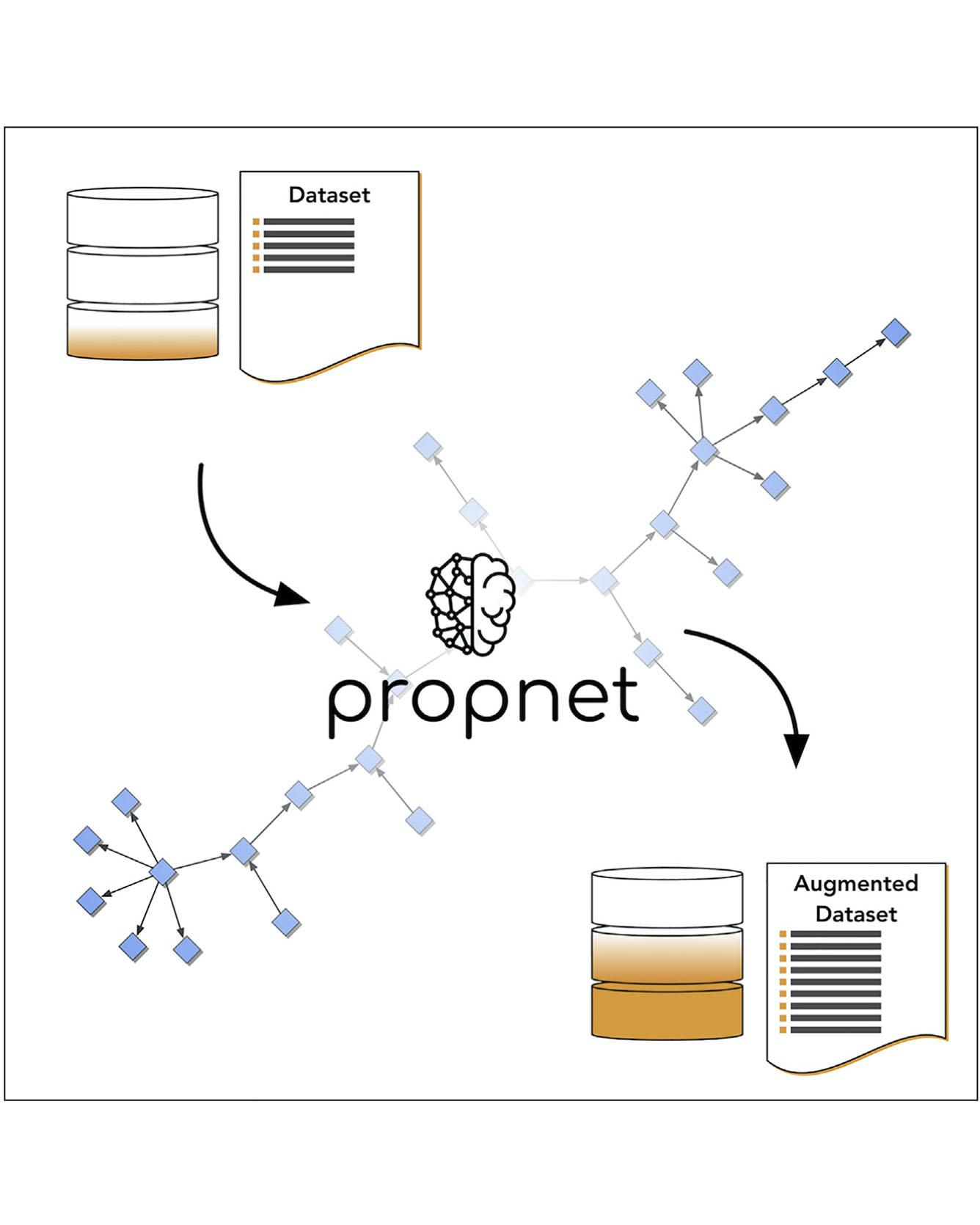
Data-driven materials science is bolstered by the recent growth of online materials databases. However, the current informatics infrastructure has yet to unlock the full knowledge available within existing datasets or to explore connections between different materials science domains. Here, we present a streamlined system for codifying and connecting materials properties in an open-source Python framework: propnet. We demonstrate the capability of this framework to augment existing datasets of materials properties: by consecutively applying a network of physical relationships to calculate related information, propnet connects disparate domain knowledge. Beyond an immediate increase in available information, the results allow for the examination of correlations between sets of properties and guide the design of multifunctional materials. By emphasizing code extensibility and simplicity, we offer this software to the materials science community for general application to any experimental or computationally derived materials database. Read More
Citations: Mrdjenovich, David, Matthew K. Horton, Joseph H. Montoya, Christian M. Legaspi, Shyam Dwaraknath, Vahe Tshitoyan, Anubhav Jain, and Kristin A. Persson. "propnet: A Knowledge Graph for Materials Science." Matter (2020).
TRI Authors: Brian Rohr, Joseph Montoya, Santosh Suram, Linda Hung
All Authors: Steven Torrisi, Matthew Carbone, Brian Rohr, Joseph Montoya, Yang Ha, Junko Yano, Santosh Suram, Linda Hung
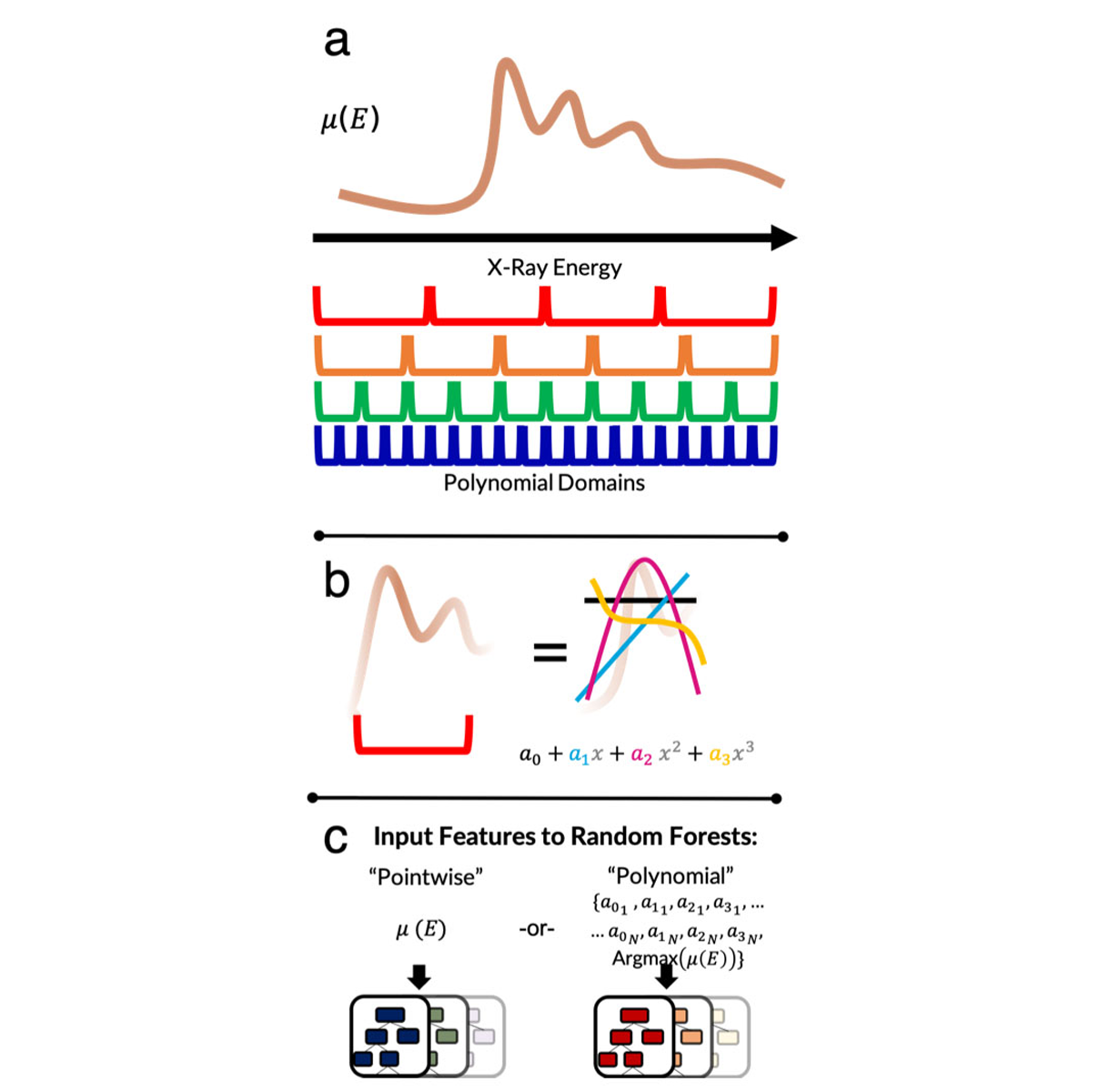
X-ray absorption spectroscopy (XAS) produces a wealth of information about the local structure of materials, but interpretation of spectra often relies on easily accessible trends and prior assumptions about the structure. Recently, researchers have demonstrated that machine learning models can automate this process to predict the coordinating environments of absorbing atoms from their XAS spectra. However, machine learning models are often difficult to interpret, making it challenging to determine when they are valid and whether they are consistent with physical theories. In this work, we present three main advances to the data-driven analysis of XAS spectra: we demonstrate the efficacy of random forests in solving two new property determination tasks (predicting Bader charge and mean nearest neighbor distance), we show that multiscale featurization can elucidate the regions and trends in spectra that encode various local properties, and we address the effect of normalization on model interpretability. The multiscale featurization transforms the spectrum into a vector of polynomial-fit features, and is contrasted with the commonly-used "pointwise" featurization that directly uses the entire spectrum as input. We find that across thousands of transition metal oxide spectra, the relative importance of features describing the curvature of the spectrum can be localized to individual energy ranges, and we can separate the importance of constant, linear, quadratic, and cubic trends, as well as the white line energy. This work has the potential to assist rigorous theoretical interpretations, expedite experimental data collection, and automate analysis of XAS spectra, thus accelerating discovery of new functional materials. Read More
Citation: Torrisi, Steven, Matthew Carbone, Brian Rohr, Joseph H. Montoya, Yang Ha, Junko Yano, Santosh Suram, and Linda Hung. "Random Forest Machine Learning Models for Interpretable X-Ray Absorption Near-Edge Structure Spectrum-Property Relationships." chemRxiv preprint (2020). doi:10.26434/chemrxiv.11873691.v1
TRI Authors: Muratahan Aykol, Santosh K. Suram* All Authors: Brian Rohr, Helge S. Stein, Dan Guevarra, Yu Wang, Joel A. Haber, Muratahan Aykol, Santosh K. Suram* and John M. Gregoire*
Sequential learning (SL) strategies, i.e. iteratively updating a machine learning model to guide experiments, have been proposed to significantly accelerate materials discovery and research. Applications on computational datasets and a handful of optimization experiments have demonstrated the promise of SL, motivating a quantitative evaluation of its ability to accelerate materials discovery, specifically in the case of physical experiments. The benchmarking effort in the present work quantifies the performance of SL algorithms with respect to a breadth of research goals: discovery of any “good” material, discovery of all “good” materials, and discovery of a model that accurately predicts the performance of new materials. To benchmark the effectiveness of different machine learning models against these goals, we use datasets in which the performance of all materials in the search space is known from high-throughput synthesis and electrochemistry experiments. Each dataset contains all pseudo-quaternary metal oxide combinations from a set of six elements (chemical space), the performance metric chosen is the electrocatalytic activity (overpotential) for the oxygen evolution reaction (OER). A diverse set of SL schemes is tested on four chemical spaces, each containing 2121 catalysts. The presented work suggests that research can be accelerated by up to a factor of 20 compared to random acquisition in specific scenarios. The results also show that certain choices of SL models are ill-suited for a given research goal resulting in substantial deceleration compared to random acquisition methods. The results provide quantitative guidance on how to tune an SL strategy for a given research goal and demonstrate the need for a new generation of materials-aware SL algorithms to further accelerate materials discovery. Read More Citation: Rohr, Brian, Helge S. Stein, Dan Guevarra, Yu Wang, Joel A. Haber, Muratahan Aykol, Santosh K. Suram, and John M. Gregoire. "Benchmarking the acceleration of materials discovery by sequential learning." Chemical Science 11, no. 10 (2020): 2696-2706.
