Featured Publications

All Publications
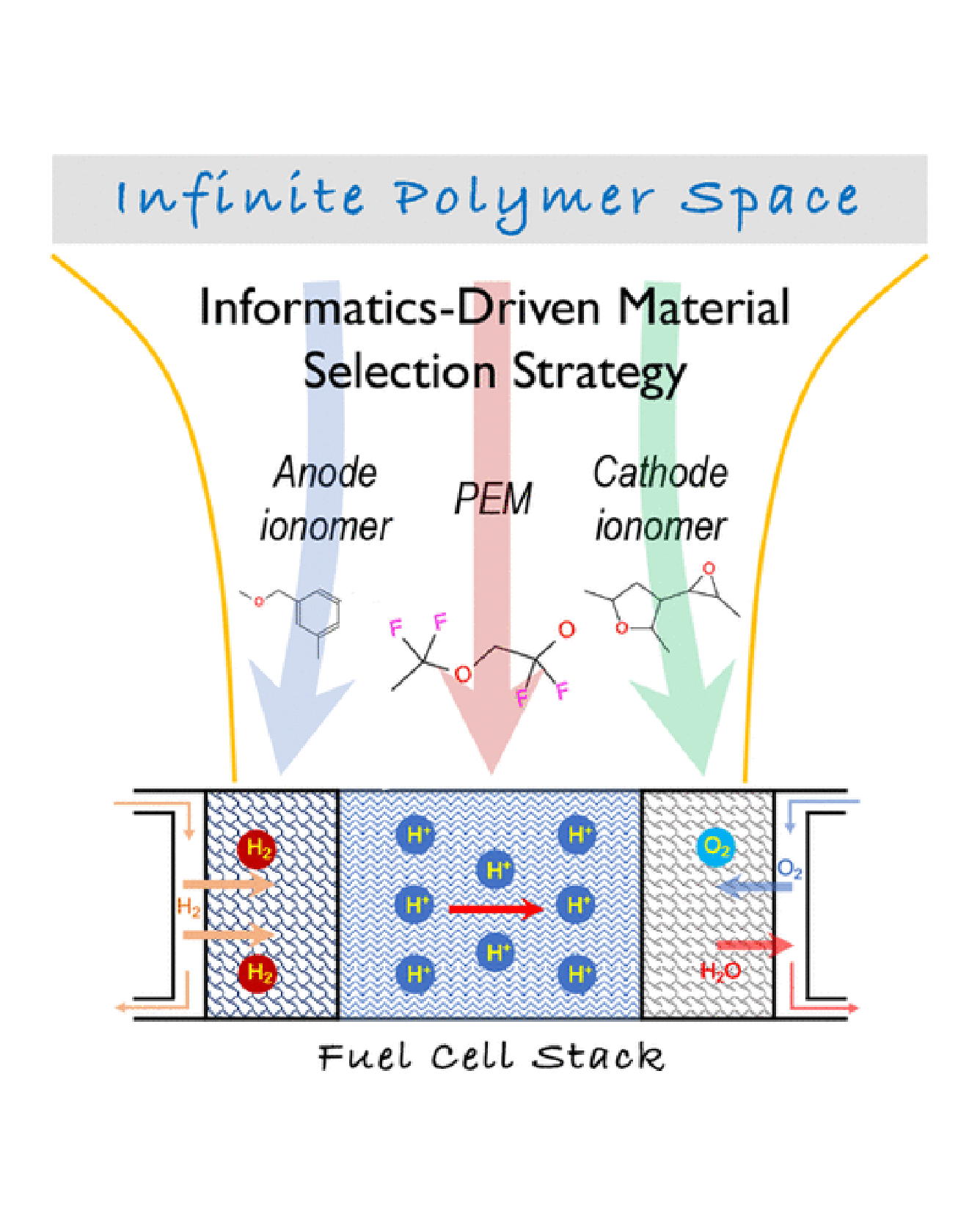
Modern fuel cell technologies use Nafion as the material of choice for the proton exchange membrane (PEM) and as the binding material (ionomer) used to assemble the catalyst layers of the anode and cathode. These applications demand high proton conductivity as well as other requirements. For example, PEM is expected to block electrons, oxygen, and hydrogen from penetrating and diffusing while the anode/cathode ionomer should allow hydrogen/oxygen to move easily, so that they can reach the catalyst nanoparticles. Given some of the well-known limits of Nafion, such as low glass-transition temperature, the community is in the midst of an active search for Nafion replacements. In this work, we present an informatics-based scheme to search large polymer chemical spaces, which includes establishing a list of properties needed for the targeted applications, developing predictive machine-learning models for these properties, defining a search space, and using the developed models to screen the search space. Using the scheme, we have identified 60 new polymer candidates for PEM, anode ionomer, and cathode ionomer that we hope will be advanced to the next step, i.e., validating the designs through synthesis and testing. The proposed informatics scheme is generic, and it can be used to select polymers for multiple applications in the future. READ MORE
Batteries are central to modern society. They are no longer just a convenience but a critical enabler of the transition to a resilient, low-carbon economy. Battery development capabilities are provided by communities spanning materials discovery, battery chemistry and electrochemistry, cell and pack design, scale-up, manufacturing, and deployments. Despite their relative maturity, data-science practices among these diverse groups are far behind the state of the art in other fields, which have demonstrated an ability to significantly improve innovation and economic impact. The negative consequences of the present paradigm include incremental improvements but few breakthroughs, significant manufacturing uncertainties, and cascading investment risks that collectively slow deployments. The primary roadblock to a battery-data-science renaissance is the requirement for large amounts of high-quality data, which are not available in the current fragmented ecosystem. Here, we identify gaps and propose principles that enable the solution by building a robust community of data hubs with standardized practices and flexible sharing options that will seed advanced tools spanning innovation to deployment. Precedents are offered that demonstrate that both public good and immense economic gains will arise from sharing valuable battery data. The proposed Battery Data Genome looks to broadly transform innovations and revolutionize their translation from research to societal impact. READ MORE
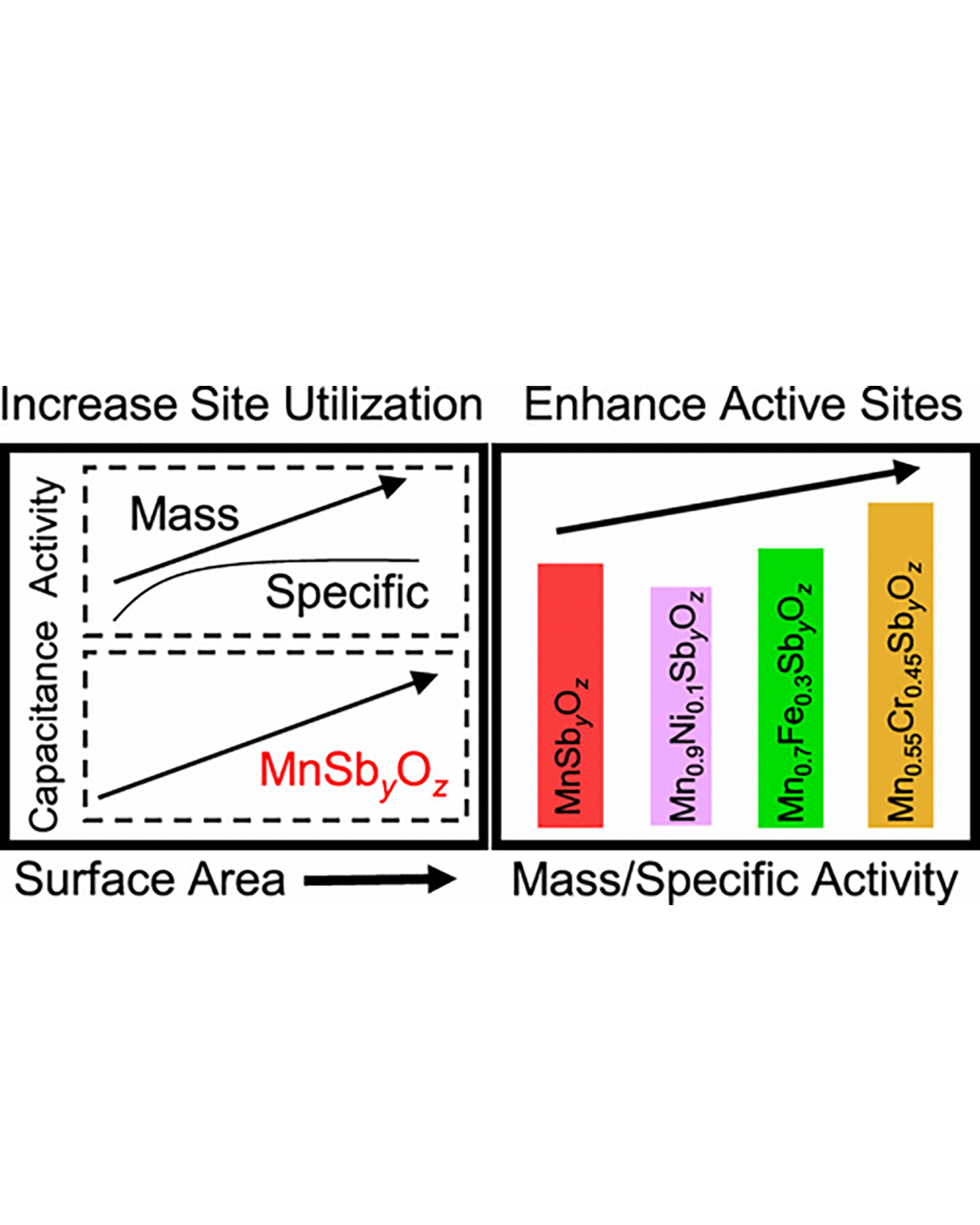
Strategies for improving the performance of nonprecious metal catalysts for the oxygen reduction reaction (ORR) can facilitate the cost-effective deployment of fuel cell devices. Electrocatalyst performance is typically improved via two approaches: increasing the number of active sites and increasing the intrinsic activity of the active site. Herein, we utilize these two methods of improving performance for MnSb2O6, which we have recently shown to be a promising ORR catalyst due to improvements in per-metal-site activity in the antimonate framework. First, electrode engineering is used to investigate the role of mass and conductive support loading in the observed ORR performance and selectivity. The apparent 2-electron selectivity is found to decrease with increases in mass and/or conductive support loading, indicating that rotating ring disk electrode studies do not necessarily measure the intrinsic selectivity of the catalyst. Second, theoretical calculations are used to identify Cr, Fe, and Ni as promising first-row transition metals for improving the intrinsic activity of MnSb2O6. Experimentally, the addition of Cr results in 3-fold and 2-fold increases in the mass and specific activities at 0.7 V vs the reversible hydrogen electrode, respectively. This enhancement is attributed to the modulation of the active site structure and Mn oxidation state with the addition of Cr. Through these studies, we gain insight into the intrinsic and extrinsic factors that govern ORR performance. READ MORE
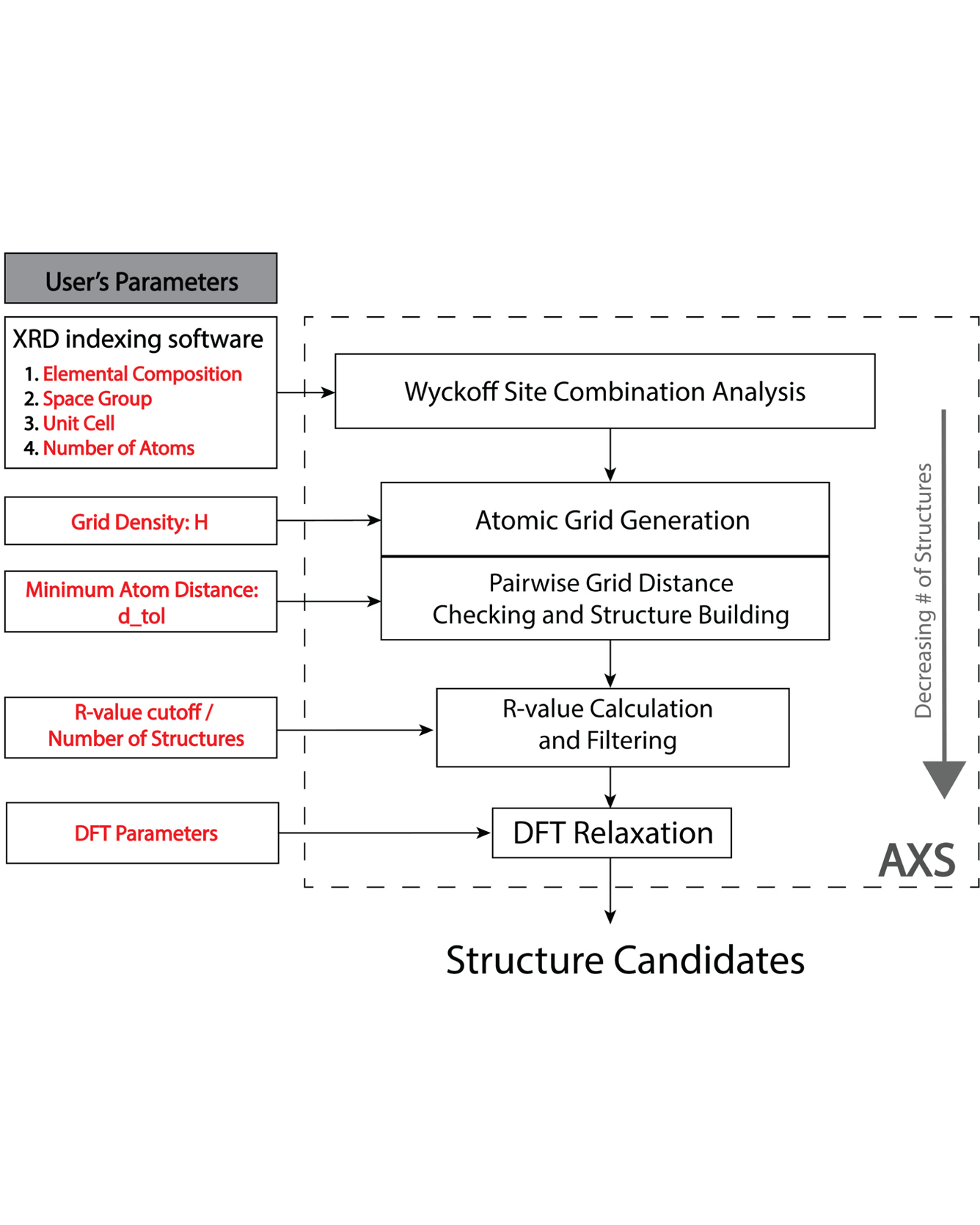
X-ray powder diffraction (XRD) is a powerful structure characterization technique, but solving unknown inorganic crystal structures from powder diffraction patterns can often be labor-intensive or speculative. We introduce an Automated XRD to Structure (AXS) solution method based on the crystal symmetry obtained from XRD patterns, an efficient search of candidate structures spanning the available degrees of freedom, and density functional theory (DFT). This methodology is completely agnostic to structural prototypes and robust in solving inorganic structures of various chemistries, crystal systems, and unit cell sizes; 92% of all crystal structures were accurately determined from the simulated XRD patterns in our benchmark set. In addition, we demonstrate the efficacy of this methodology on experimental XRD patterns by solving the crystal structures of Li8Hf6O8, Li3CrO4, and LiFeO2. READ MORE
Density functional theory (DFT) has been widely applied in modern materials discovery and many materials databases, including the open quantum materials database (OQMD), contain large collections of calculated DFT properties of experimentally known crystal structures and hypothetical predicted compounds. Since the beginning of the OQMD in late 2010, over one million compounds have now been calculated and stored in the database, which is constantly used by worldwide researchers in advancing materials studies. The growth of the OQMD depends on project-based high-throughput DFT calculations, including structure-based projects, property-based projects, and most recently, machine-learning-based projects. Another major goal of the OQMD is to ensure the openness of its materials data to the public and the OQMD developers are constantly working with other materials databases to reach a universal querying protocol in support of the FAIR data principles. READ MORE

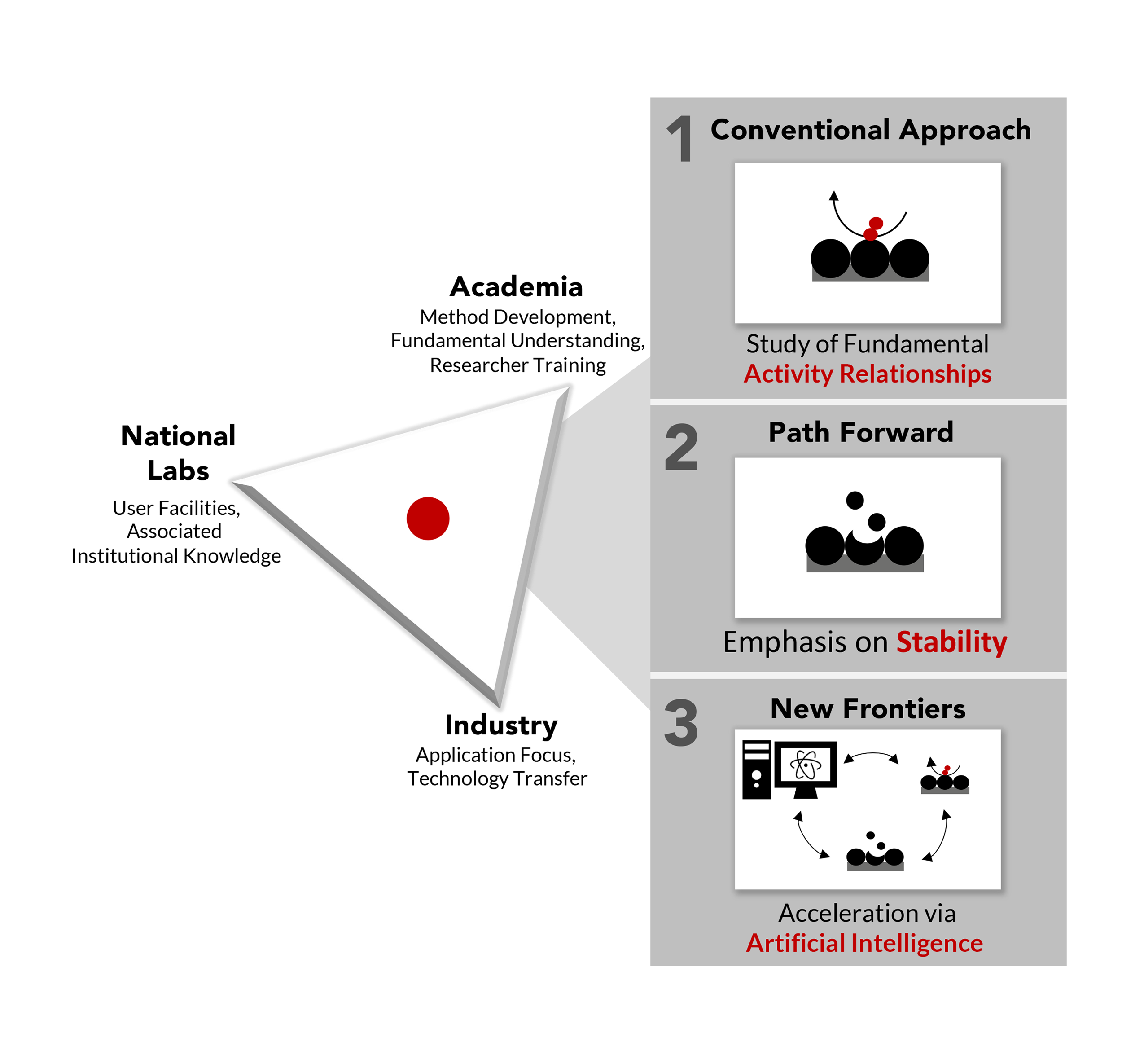
In this perspective, we highlight results of a research consortium devoted to advancing understanding of oxygen reduction reaction (ORR) catalysis as a means to inform fuel cell science. We demonstrate how targeted collaborations between different institutions from academic, national lab, and industry backgrounds and different scientific disciplines like theory, experiment, and characterization can yield unique insights into fuel cell catalysts. We comment on such insights into material designs for platinum-group-metal alloys, transition metal oxides, and non-traditional materials including metal-organic frameworks; systems that have served as the foundational building blocks for our consortium. We also motivate a renewed focus on catalyst durability in light of emerging technological requirements and paths forward in understanding in situ and operando electrochemical stability. Finally, we describe new frontiers ORR research can take and how emerging artificial intelligence tools can assist researchers in capturing data, selecting new experiments, and guiding characterization to accelerate the design and discovery of fuel cell catalysts. A main goal of sharing this perspective is to discuss the rationale for our future research plans based on our consortium work. However, we also hope to illustrate both the potential impact of a collaborative strategy with the hopes of inspiring a higher degree of Industry-Academia-National Laboratory collaboration and encourage other centers and consortiums to distill and share their findings in a similar perspective-type article. Together we hope to enable the fuel cell research community to engage in a discussion of strategies for research and accelerated development of catalysts with improved activity and stability. READ MORE
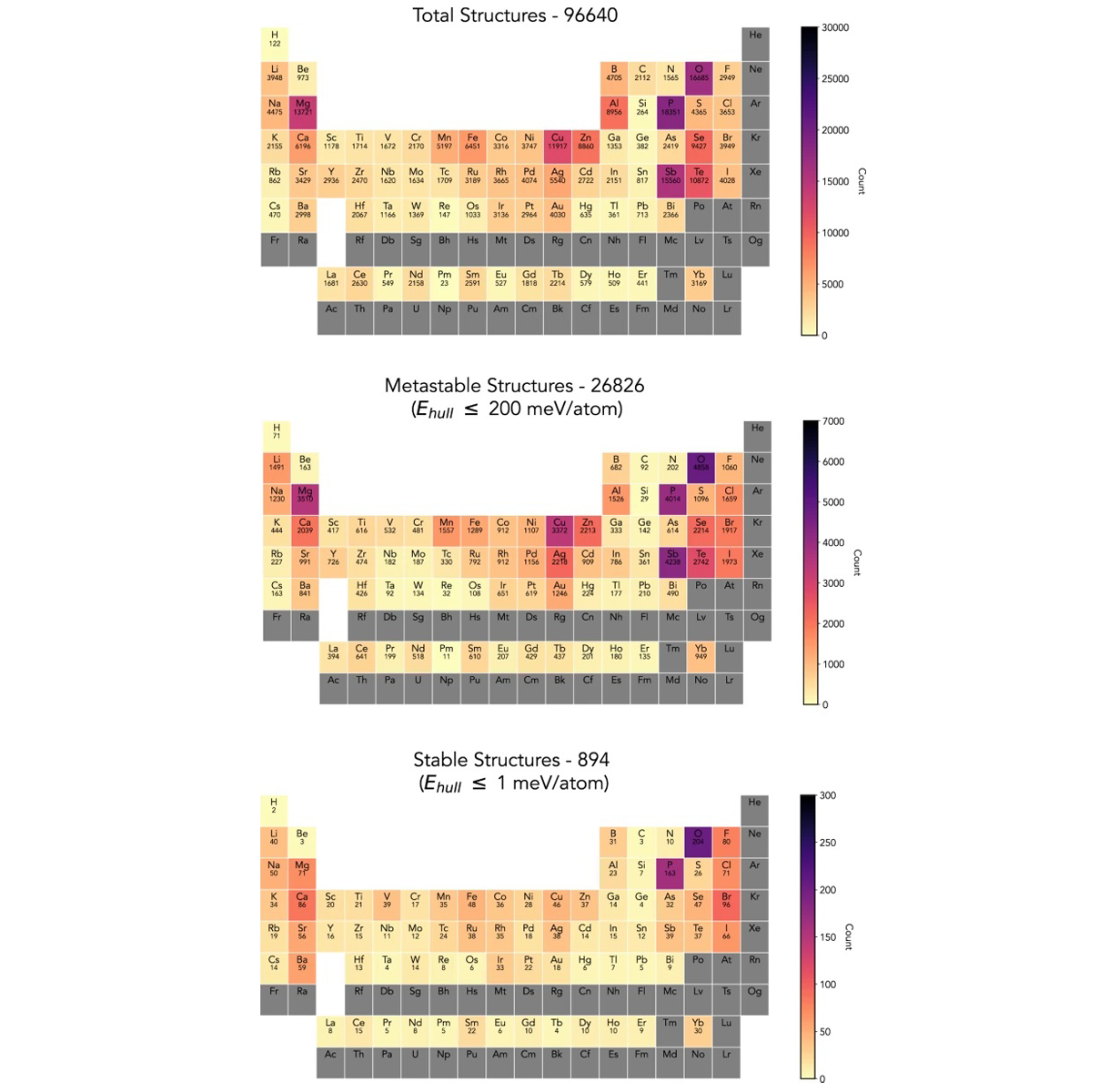
We report a dataset of 96640 crystal structures discovered and computed using our previously published autonomous, density functional theory (DFT) based, active-learning workflow named CAMD (Computational Autonomy for Materials Discovery). Of these, 894 are within 1 meV/atom of the convex hull and 26826 are within 200 meV/atom of the convex hull. The dataset contains DFT-optimized pymatgen crystal structure objects, DFT-computed formation energies and phase stability calculations from the convex hull. It contains a variety of spacegroups and symmetries derived from crystal prototypes derived from known experimental compounds, and was generated from active learning campaigns of various chemical systems. This dataset can be used to benchmark future active-learning or generative efforts for structure prediction, to seed new efforts of experimental crystal structure discovery, or to construct new models of structure-property relationships. READ MORE
The growing field of data-driven materials research poses a challenge to educators: teaching machine learning to materials scientists. We share our recent experiences and lessons learnt from organizing educational sessions at the fall 2021 meeting of the Materials Research Society. READ MORE

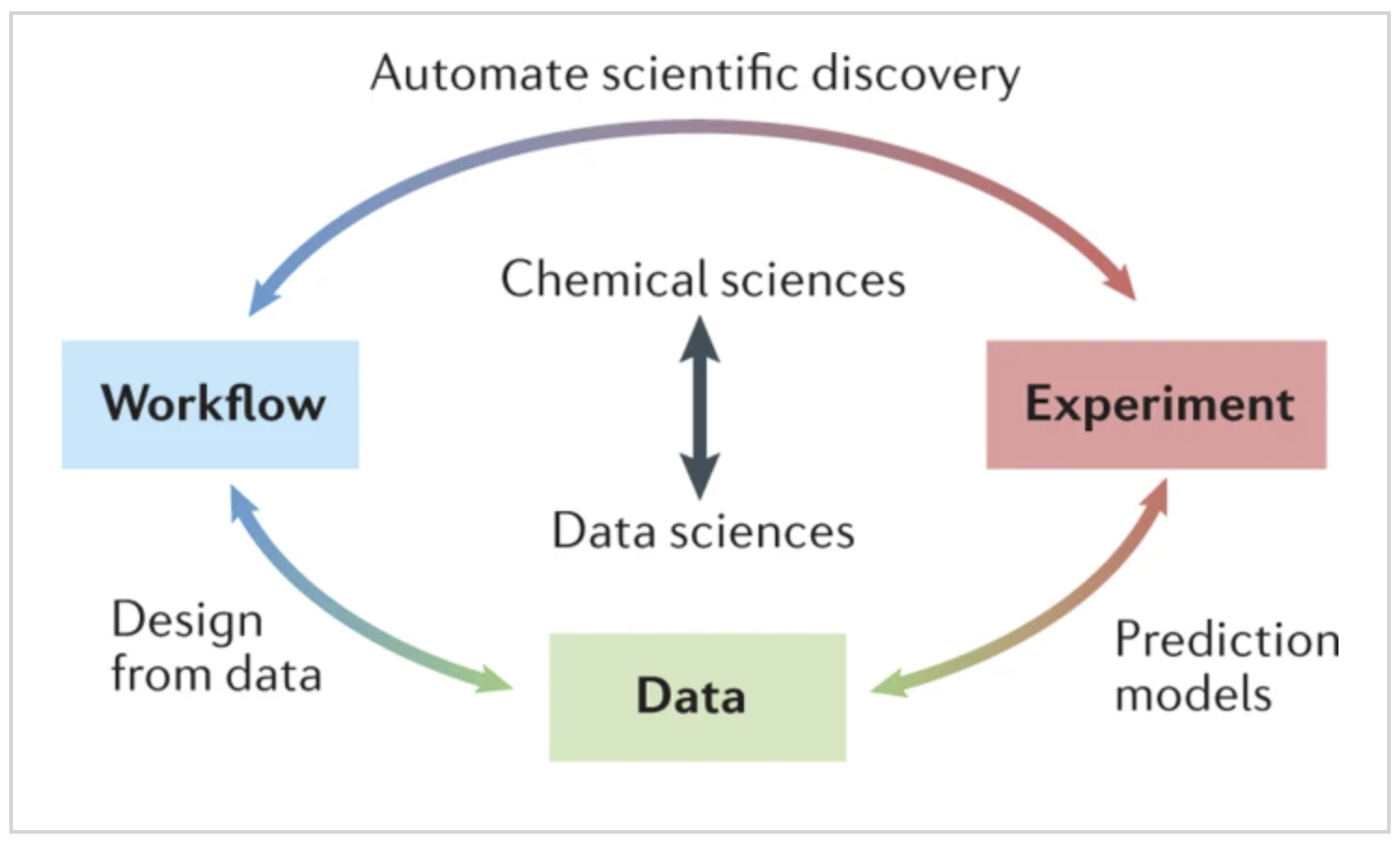
The physical sciences community is increasingly taking advantage of the possibilities offered by modern data science to solve problems in experimental chemistry and potentially to change the way we design, conduct and understand results from experiments. Successfully exploiting these opportunities involves considerable challenges. In this Expert Recommendation, we focus on experimental co-design and its importance to experimental chemistry. We provide examples of how data science is changing the way we conduct experiments, and we outline opportunities for further integration of data science and experimental chemistry to advance these fields. Our recommendations include establishing stronger links between chemists and data scientists; developing chemistry-specific data science methods; integrating algorithms, software and hardware to ‘co-design’ chemistry experiments from inception; and combining diverse and disparate data sources into a data network for chemistry research. READ MORE
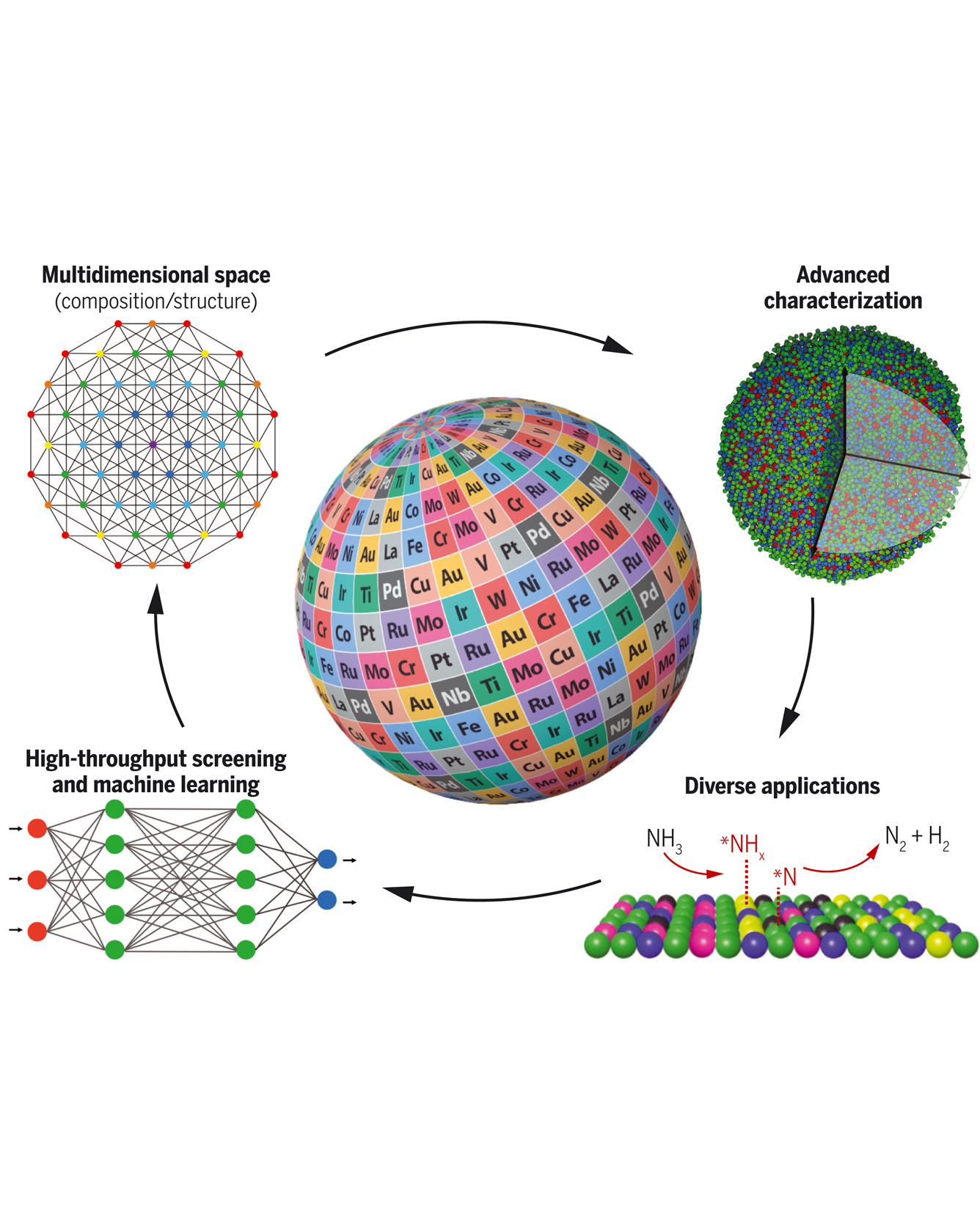
High-entropy nanoparticles have become a rapidly growing area of research in recent years. Because of their multielemental compositions and unique high-entropy mixing states (i.e., solid-solution) that can lead to tunable activity and enhanced stability, these nanoparticles have received notable attention for catalyst design and exploration. However, this strong potential is also accompanied by grand challenges originating from their vast compositional space and complex atomic structure, which hinder comprehensive exploration and fundamental understanding. Through a multidisciplinary view of synthesis, characterization, catalytic applications, high-throughput screening, and data-driven materials discovery, this review is dedicated to discussing the important progress of high-entropy nanoparticles and unveiling the critical needs for their future development for catalysis, energy, and sustainability applications. READ MORE