Featured Publications

All Publications
Natural Language Processing (NLP), a cornerstone field within artificial intelligence, has been increasingly utilized in the field of materials science literature. Our study conducts a reproducibility analysis of two pioneering works within this domain: "Machine-learned and codified synthesis parameters of oxide materials" by Kim et al., and "Unsupervised word embeddings capture latent knowledge from materials science literature" by Tshitoyan et al. We aim to comprehend these studies from a reproducibility perspective, acknowledging their significant influence on the field of materials informatics, rather than critiquing them. Our study indicates that both papers offered thorough workflows, tidy and well-documented codebases, and clear guidance for model evaluation. This makes it easier to replicate their results successfully and partially reproduce their findings. In doing so, they set commendable standards for future materials science publications to aspire to. However, our analysis also highlights areas for improvement such as to provide access to training data where copyright restrictions permit, more transparency on model architecture and the training process, and specifications of software dependency versions. We also cross-compare the word embedding models between papers, and find that some key differences in reproducibility and cross-compatibility are attributable to design choices outside the bounds of the models themselves. In summary, our study appreciates the benchmark set by these seminal papers while advocating for further enhancements in research reproducibility practices in the field of NLP for materials science. This balance of understanding and continuous improvement will ultimately propel the intersecting domains of NLP and materials science literature into a future of exciting discoveries. READ MORE
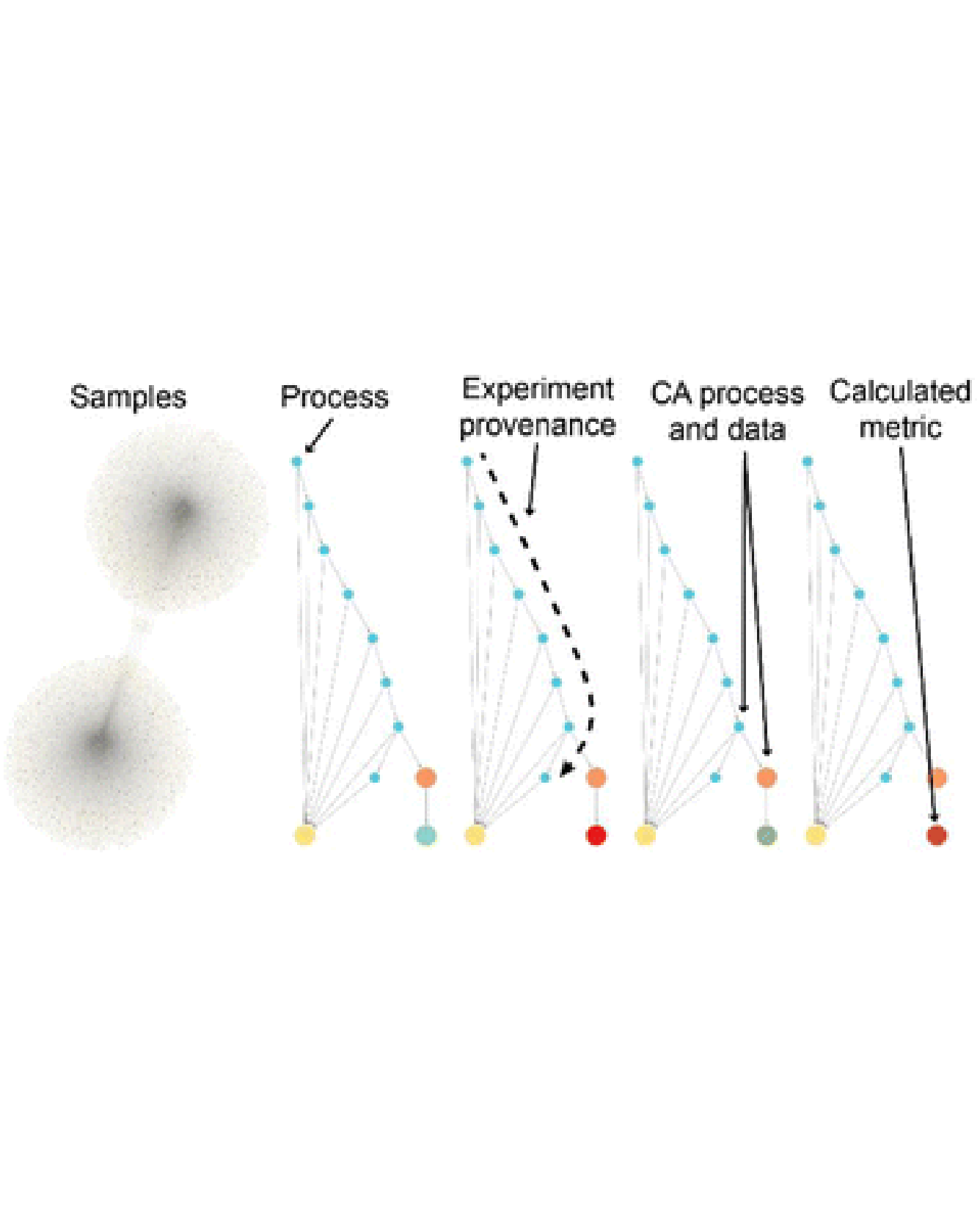
Materials knowledge is inherently hierarchical. While high-level descriptors such as composition and structure are valuable for contextualizing materials data, the data must ultimately be considered in the context of its low-level acquisition details. Graph databases offer an opportunity to represent hierarchical relationships among data, organizing semantic relationships into a knowledge graph. Herein, we establish a knowledge graph of materials experiments whose construction encodes the complete provenance of each material sample and its associated experimental data and metadata. Additional relationships among materials and experiments further encode knowledge and facilitate data exploration. We illustrate the Materials Experiment Knowledge Graph (MekG) using several use cases, demonstrating the value of modern graph databases for the enterprise of data-driven materials science. READ MORE
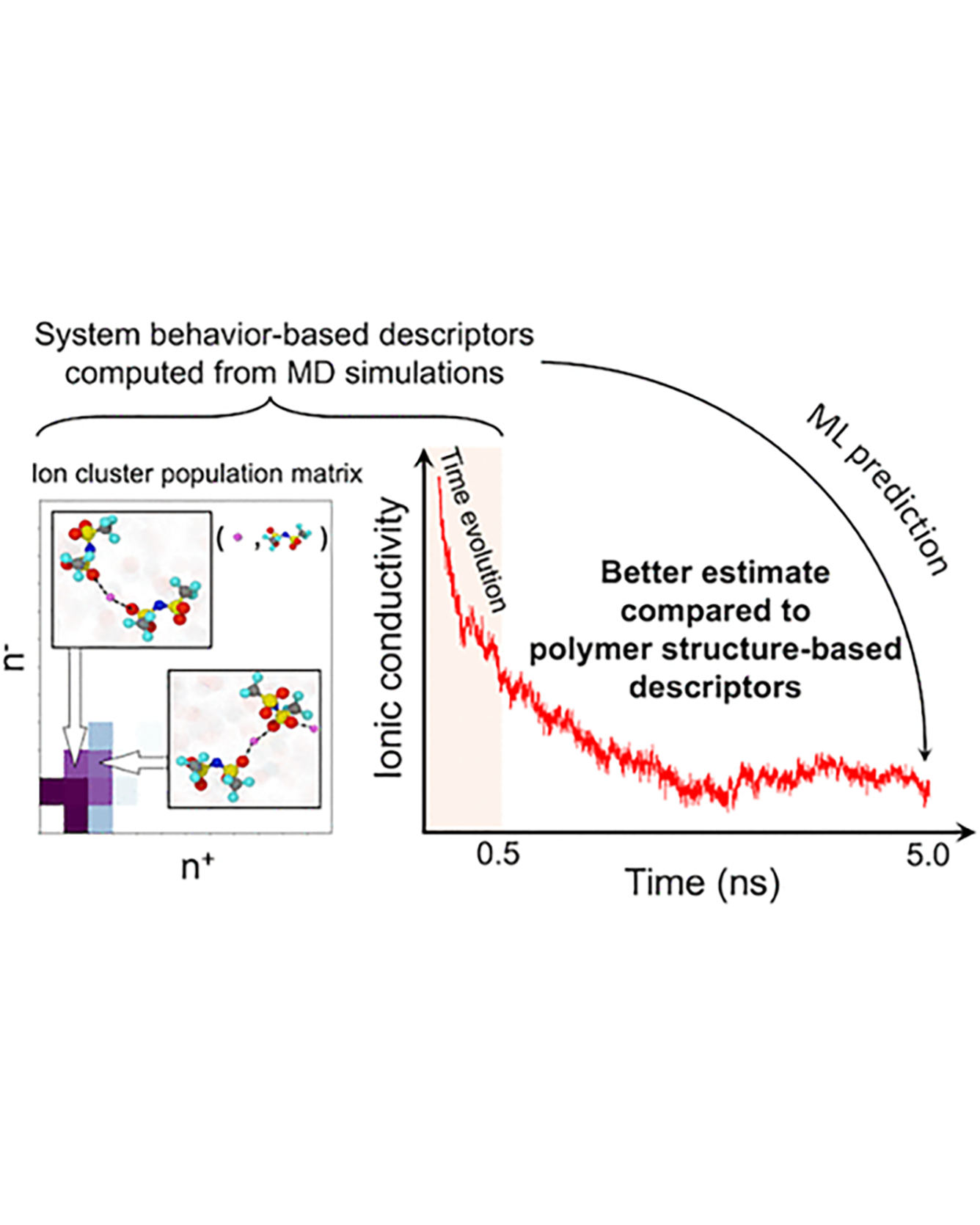
Molecular dynamics simulations are useful tools to screen solid polymer electrolytes with suitable properties applicable to Li-ion batteries. However, due to the vast design space of polymers, it is highly desirable to accelerate the screening by reducing the computational time of ion transport properties from simulations. In this study, we show that with a judicious choice of descriptors we can predict the equilibrium ion transport properties in LiTFSI–homopolymer systems within the first 0.5 ns of the production run of simulations. Specifically, we find that descriptors that include information about the behavior of the system, such as ion clustering and time evolution of ion transport properties, have several advantages over polymer structure-based descriptors, as they encode system (polymer and salt) behavior rather than just the class of polymers and can be computed at any time point during the simulations. These characteristics increase the applicability of our descriptors to a wide range of polymer systems (e.g., copolymers, blend of polymers, salt concentrations, and temperatures) and can be impactful in significantly shortening the discovery pipeline for solid polymer electrolytes. READ MORE
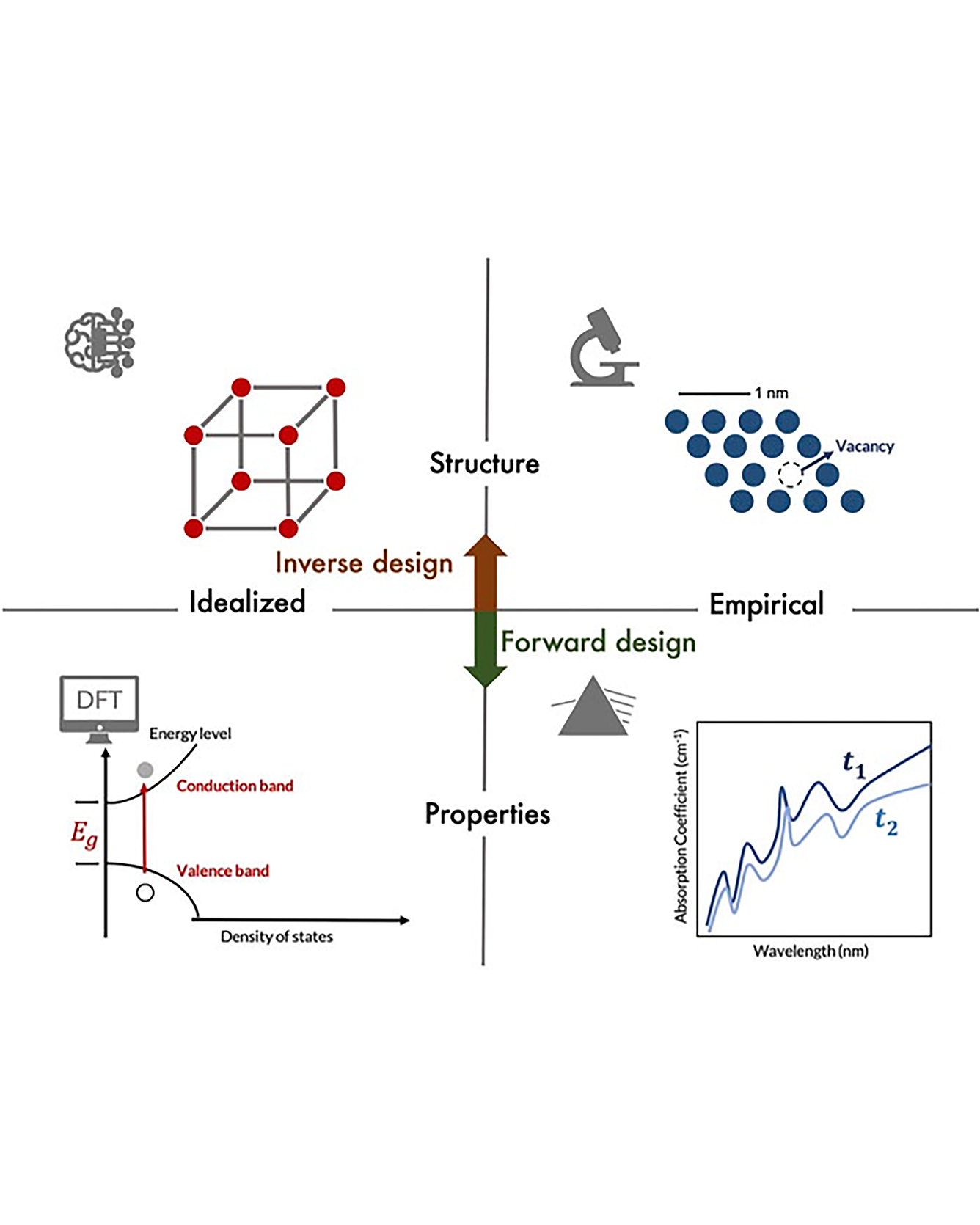
Machine learning (ML) is gaining popularity as a tool for materials scientists to accelerate computation, automate data analysis, and predict materials properties. The representation of input material features is critical to the accuracy, interpretability, and generalizability of data-driven models for scientific research. In this Perspective, we discuss a few central challenges faced by ML practitioners in developing meaningful representations, including handling the complexity of real-world industry-relevant materials, combining theory and experimental data sources, and describing scientific phenomena across timescales and length scales. We present several promising directions for future research: devising representations of varied experimental conditions and observations, the need to find ways to integrate machine learning into laboratory practices, and making multi-scale informatics toolkits to bridge the gaps between atoms, materials, and devices. READ MORE
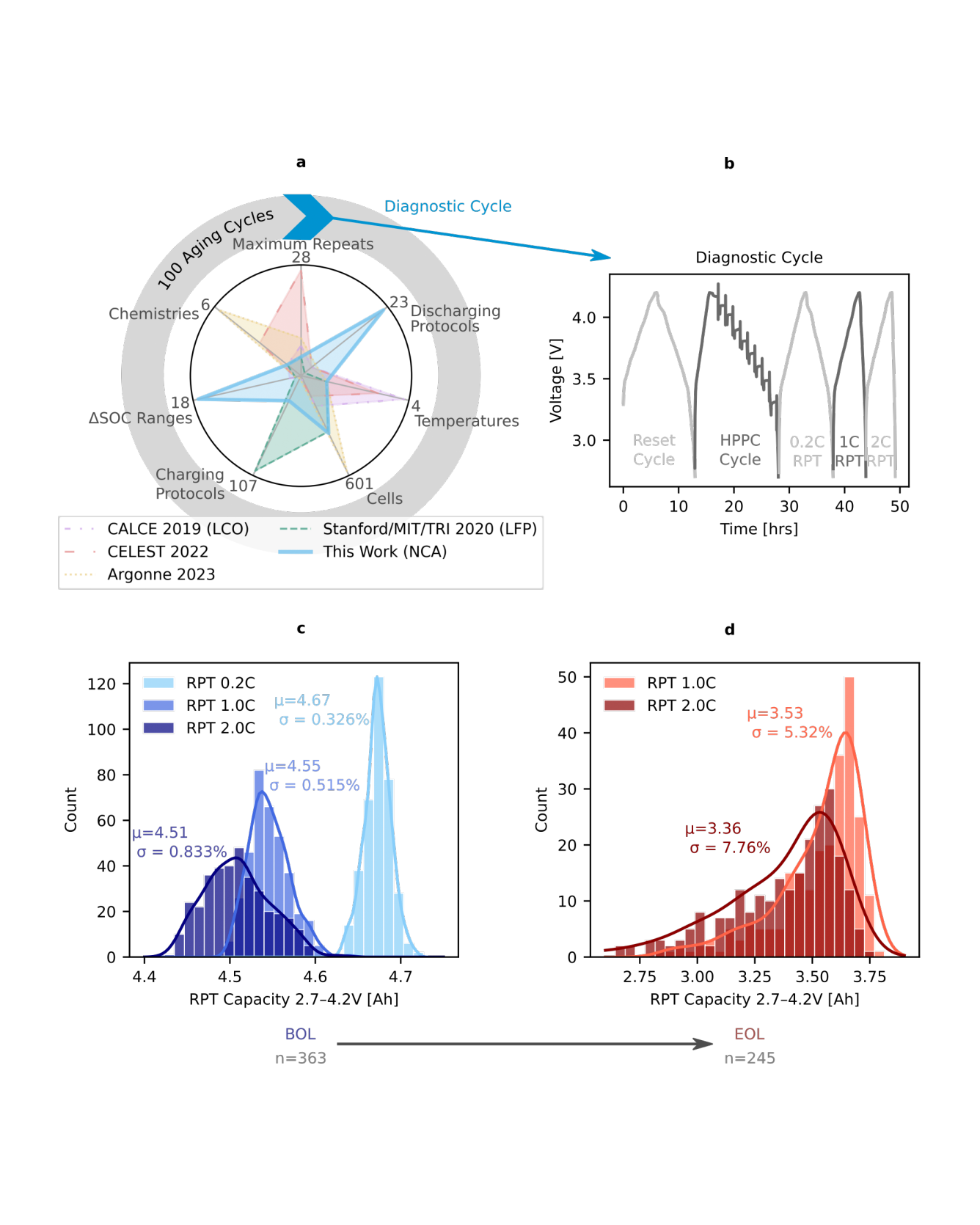
To reliably deploy lithium-ion batteries, a fundamental understanding of cycling and aging behavior is critical. Battery aging, however, consists of complex and highly coupled phenomena, making it challenging to develop a holistic interpretation. In this work, we generate a diverse battery cycling dataset with a broad range of degradation trajectories, consisting of 363 high energy density commercial Li(Ni,Co,Al)O$_2$/Graphite + SiO$_x$ cylindrical 21700 cells cycled under 218 unique cycling protocols. We consolidate aging via 16 mechanistic state-of-health (SOH) metrics, including cell-level performance metrics, electrode-specific capacities/state-of-charges (SOCs), and aging trajectory descriptors. Through the use of interpretable machine learning and explainable features, we deconvolute the underlying factors that contribute to battery degradation. This generalizable data-driven framework reveals the complex interplay between cycling conditions, degradation modes, and SOH, representing a holistic approach towards understanding battery aging. READ MORE
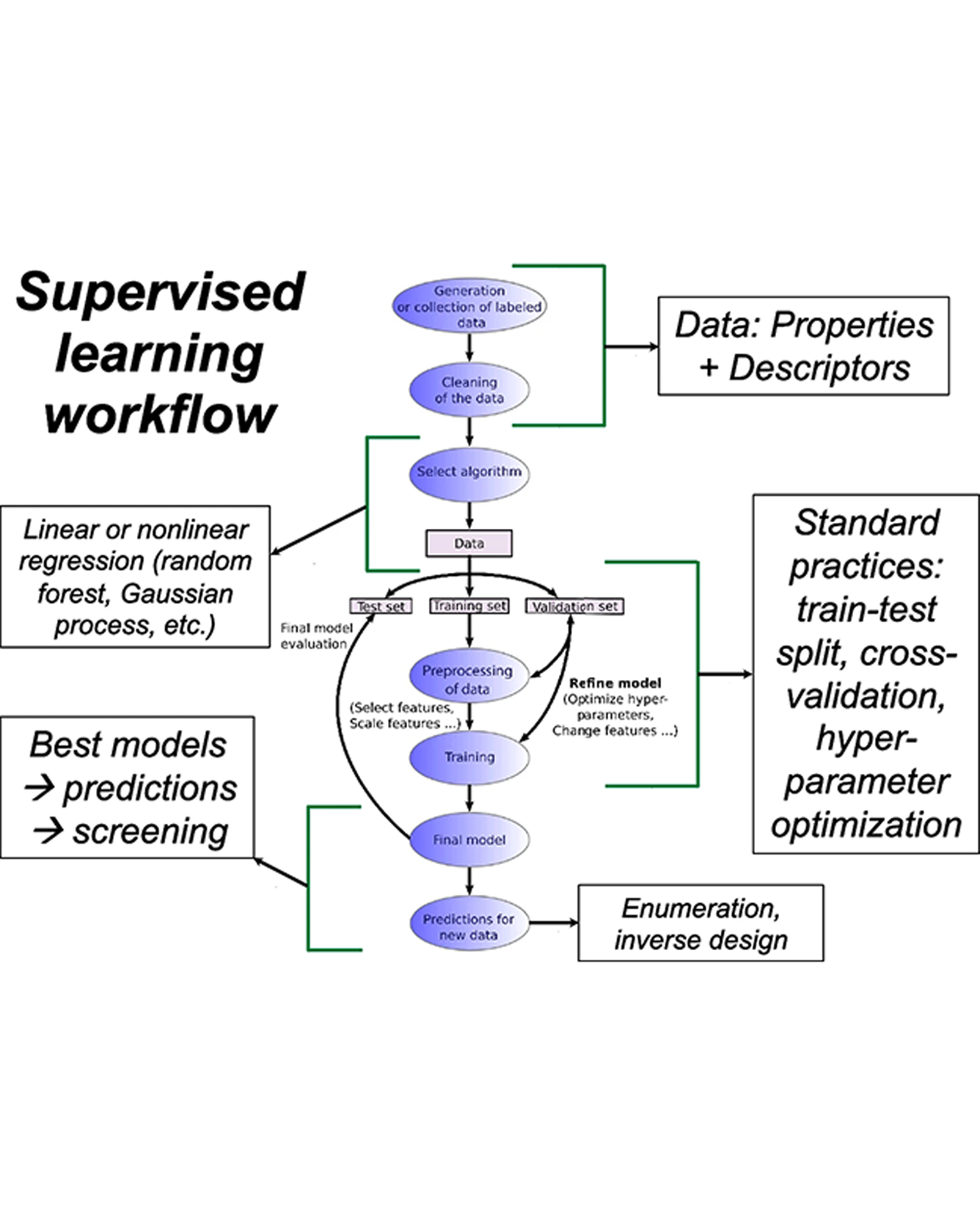
The burgeoning field of materials informatics necessitates a focus on educating the next generation of materials scientists in the concepts of data science, artificial intelligence (AI), and machine learning (ML). In addition to incorporating these topics in undergraduate and graduate curricula, regular hands-on workshops present the most effective medium to initiate researchers to informatics and have them start applying the best AI/ML tools to their own research. With the help of the Materials Research Society (MRS), members of the MRS AI Staging Committee, and a dedicated team of instructors, we successfully conducted workshops covering the essential concepts of AI/ML as applied to materials data, at both the Spring and Fall Meetings in 2022, with plans to make this a regular feature in future meetings. In this article, we discuss the importance of materials informatics education via the lens of these workshops, including details such as learning and implementing specific algorithms, the crucial nuts and bolts of ML, and using competitions to increase interest and participation. READ MORE
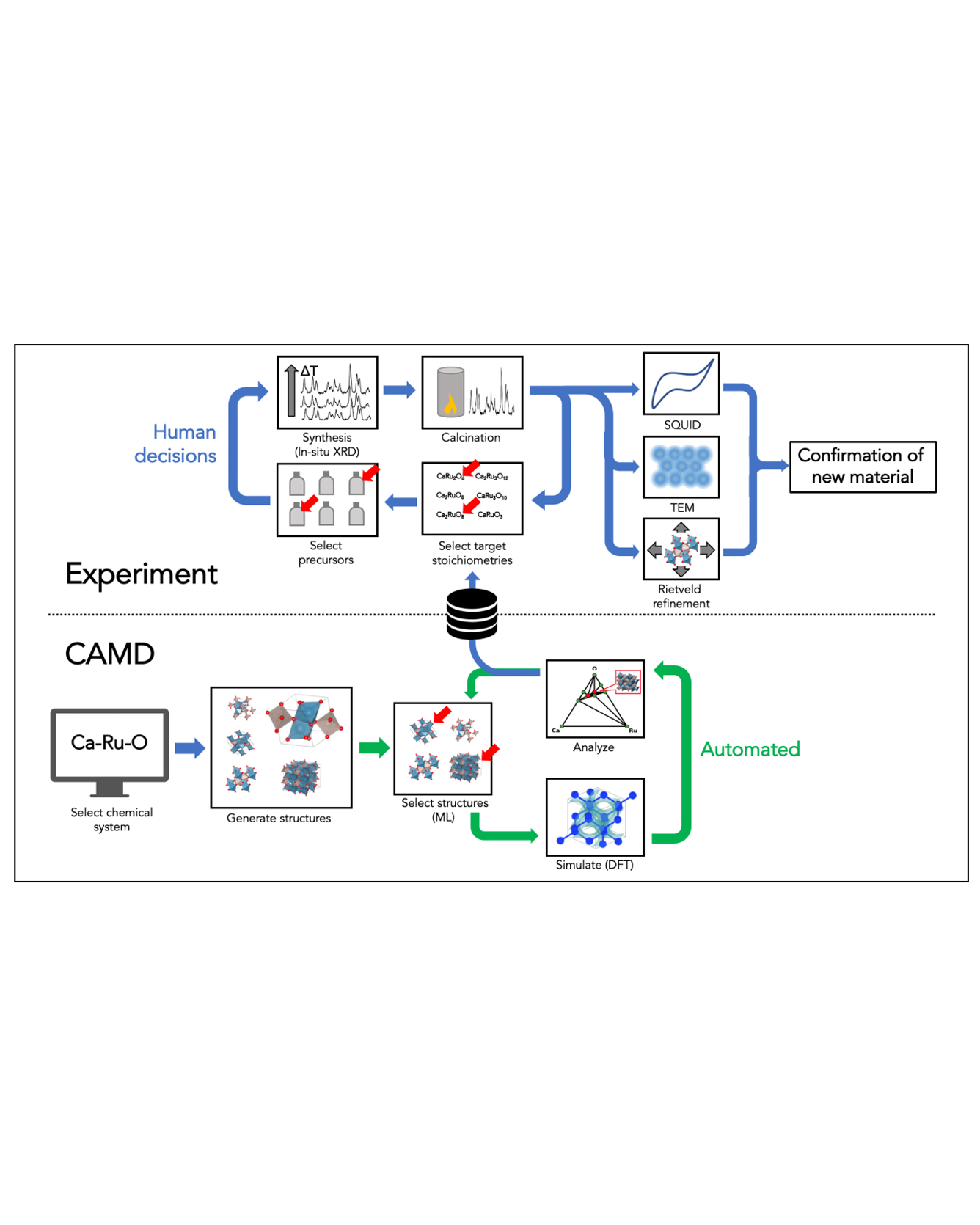
Exploratory synthesis has been the main generator of new inorganic materials for decades. However, our Edisonian and bias-prone processes of synthetic exploration alone are no longer sufficient in an age that demands rapid advances in materials development. In this work, we demonstrate one of the first end-to-end attempts towards systematic, computer-aided discovery and laboratory synthesis of inorganic crystalline compounds as a modern alternative to purely exploratory synthesis. Our approach initializes materials discovery campaigns by autonomously mapping the synthetic feasibility of a chemical system using density functional theory with AI feedback. Following expert-driven down-selection of newly generated phases, we use solid-state synthesis and in situ characterization via hot-stage X-ray diffraction in order to realize new ternary oxide phases experimentally. We applied this strategy in six ternary transition-metal oxide chemistries previously considered well-explored, one of which culminated in the discovery of two novel phases of calcium ruthenates. Detailed characterization using room temperature X-ray powder diffraction, 4D-STEM and SQUID measurements identify the structure, composition and confirm distinct properties, including distinct defect concentrations, of one of the new phases formed in our experimental campaigns. While the discovery of a new material guided by AI and DFT theory represents a milestone, our procedure and results also highlight a number of critical gaps in the process that can inform future efforts towards the improvement of AI-coupled methodologies, which are discussed. READ MORE
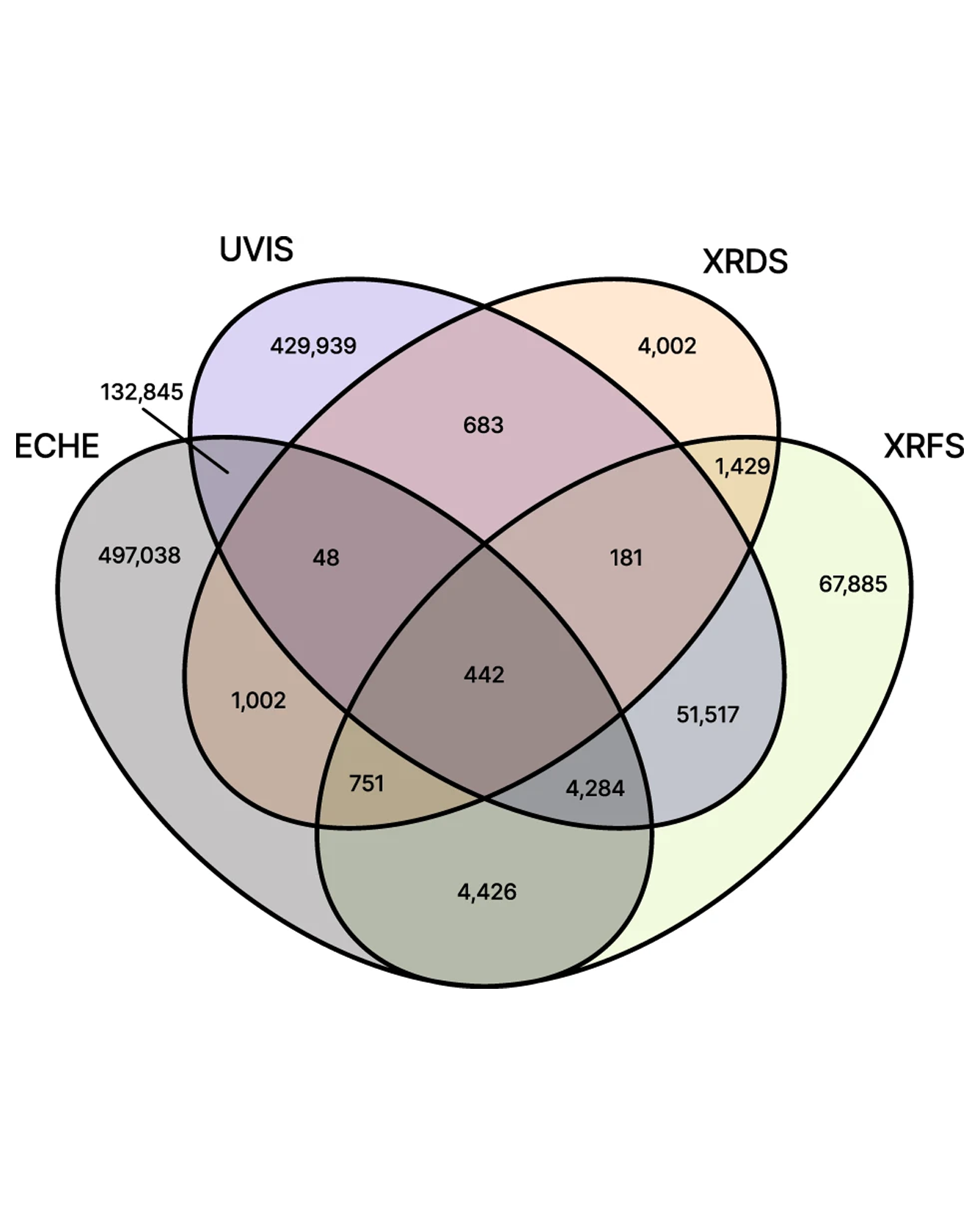
We present a database resulting from high throughput experimentation, primarily on metal oxide solid state materials. The central relational database, the Materials Provenance Store (MPS), manages the metadata and experimental provenance from acquisition of raw materials, through synthesis, to a broad range of materials characterization techniques. Given the primary research goal of materials discovery of solar fuels materials, many of the characterization experiments involve electrochemistry, along with optical, structural, and compositional characterizations. The MPS is populated with all information required for executing common data queries, which typically do not involve direct query of raw data. The result is a database file that can be distributed to users so that they can independently execute queries and subsequently download the data of interest. We propose this strategy as an approach to manage the highly heterogeneous and distributed data that arises from materials science experiments, as demonstrated by the management of over 30 million experiments run on over 12 million samples in the present MPS release. READ MORE
Crystal Toolkit is an open source tool for viewing, analyzing and transforming crystal structures, molecules and other common forms of materials science data in an interactive way. It is intended to help beginners rapidly develop web-based apps to explore their own data or to help developers make their research algorithms accessible to a broader audience of scientists who might not have any training in computer programming and who would benefit from graphical interfaces. Crystal Toolkit comes with a library of ready-made components that can be assembled to make complex web apps: simulation of powder and single crystalline diffraction patterns, convex hull phase diagrams, Pourbaix diagrams, electronic band structures, analysis of local chemical environments and symmetry, and more. Crystal Toolkit is now powering the Materials Project website frontend, providing user-friendly access to its database of computed materials properties. In the future, it is hoped that new visualizations might be prototyped using Crystal Toolkit to help explore new forms of data being generated by the materials science community, and that this in turn can help new materials scientists develop intuition for how their data behaves and the insights that might be found within. Crystal Toolkit will remain a work-in-progress and is open to contributions from the community. READ MORE
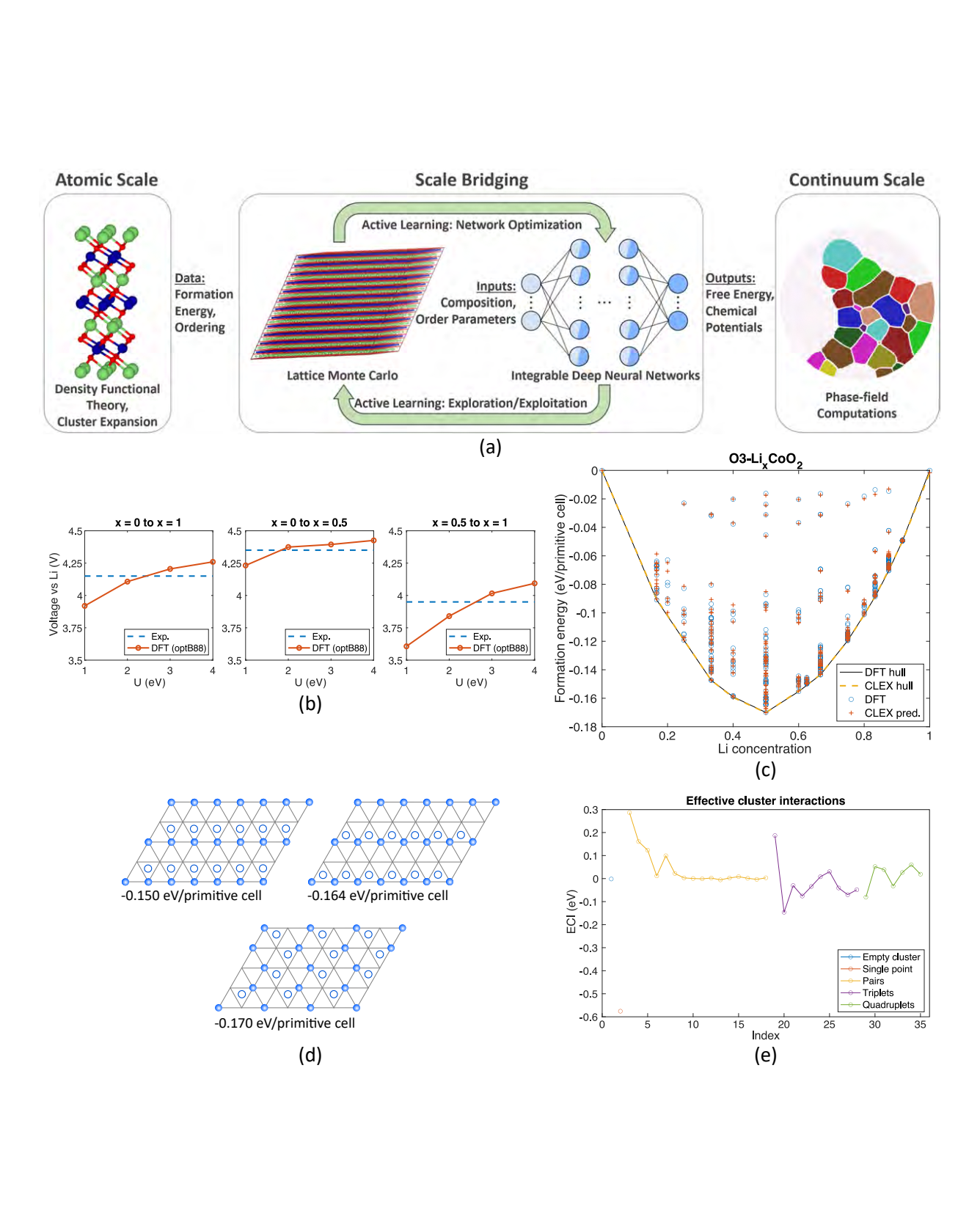
LixTMO2 (TM=Ni, Co, Mn) forms an important family of cathode materials for Li-ion batteries, whose performance is strongly governed by Li composition-dependent crystal structure and phase stability. Here, we use LixCoO2 (LCO) as a model system to benchmark a machine learning-enabled framework for bridging scales in materials physics. We focus on two scales: (a) assemblies of thousands of atoms described by density functional theory-informed statistical mechanics, and (b) continuum phase field models to study the dynamics of order-disorder transitions in LCO. Central to the scale bridging is the rigorous, quantitatively accurate, representation of the free energy density and chemical potentials of this material system by coarsegraining formation energies for specific atomic configurations. We develop active learning workflows to train recently developed integrable deep neural networks for such high-dimensional free energy density and chemical potential functions. The resulting, first principles-informed, machine learning-enabled, phase-field computations allow us to study LCO cathodes' phase evolution in terms of temperature, morphology, charge cycling and particle size. READ MORE