Featured Publications

All Publications
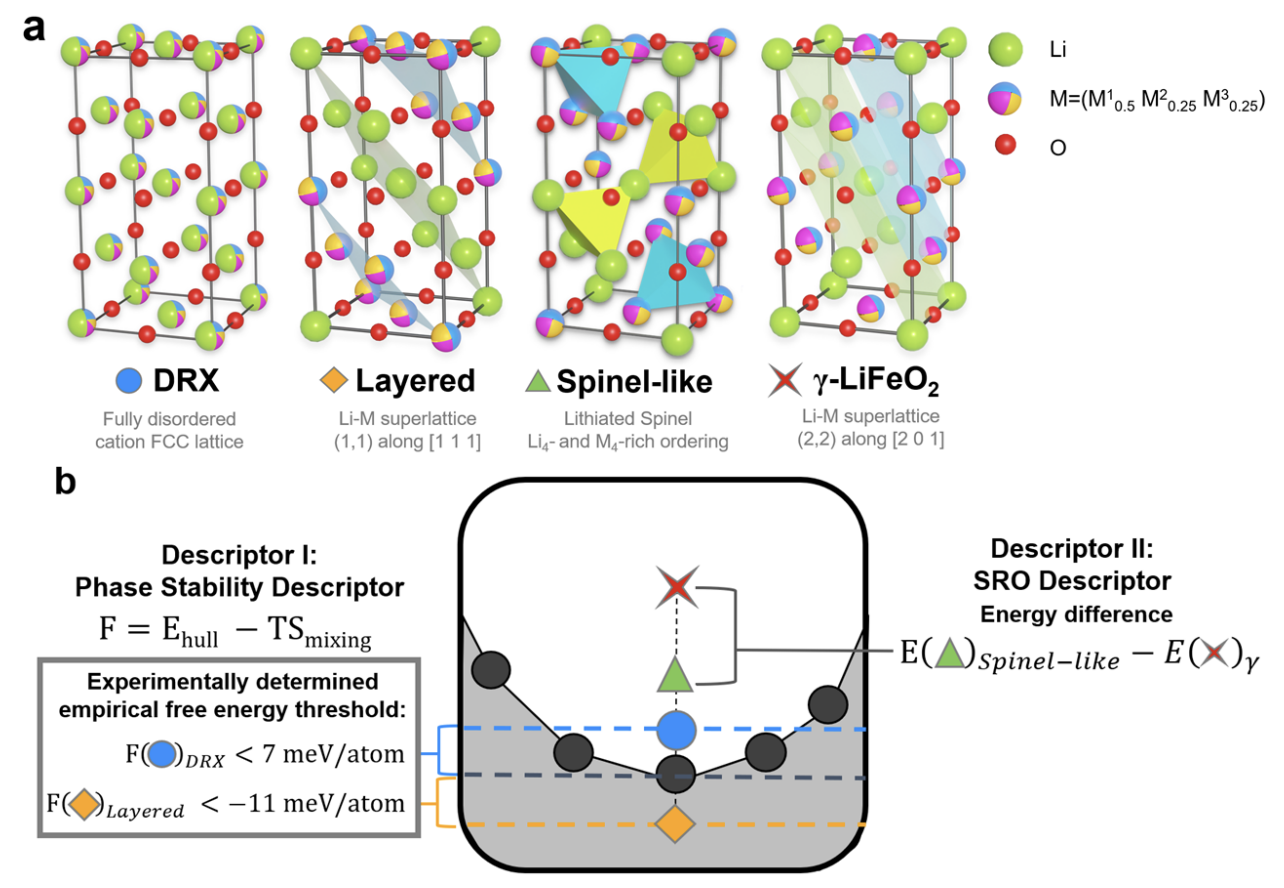
Newly designed Li-ion battery cathode materials with high capacity and greater flexibility in chemical composition will be critical for the growing electric vehicles market. Cathode structures with cation disorder were once considered suboptimal, but recent demonstrations have highlighted their potential in Li1+xM1−xO2 chemistries with a wide range of metal combinations M. By relaxing the strict requirements of maintaining ordered Li diffusion pathways, countless multi-metal compositions in LiMO2 may become viable, aiding the quest for high-capacity cobalt-free cathodes. A challenge presented by this freedom in composition space is designing compositions which possess specific, tailored types of both long- and short-range orderings, which can ensure both phase stability and Li diffusion. However, the combinatorial complexity associated with local cation environments impedes the development of general design guidelines for favorable orderings. Here we propose ordering design frameworks from computational ordering descriptors, which in tandem with low-cost heuristics and elemental statistics can be used to simultaneously achieve compositions that possess favorable phase stability as well as configurations amenable to Li diffusion. Utilizing this computational framework, validated through multiple successful synthesis and characterization experiments, we not only demonstrate the design of LiCr0.75Fe0.25O2, showcasing initial charge capacity of 234 mAhg−1 and 320 mAhg−1 in its 20% Li-excess variant Li1.2Cr0.6Fe0.2O2, but also present the elemental ordering statistics for 32 elements, informed by one of the most extensive first-principles studies of ordering tendencies known to us.
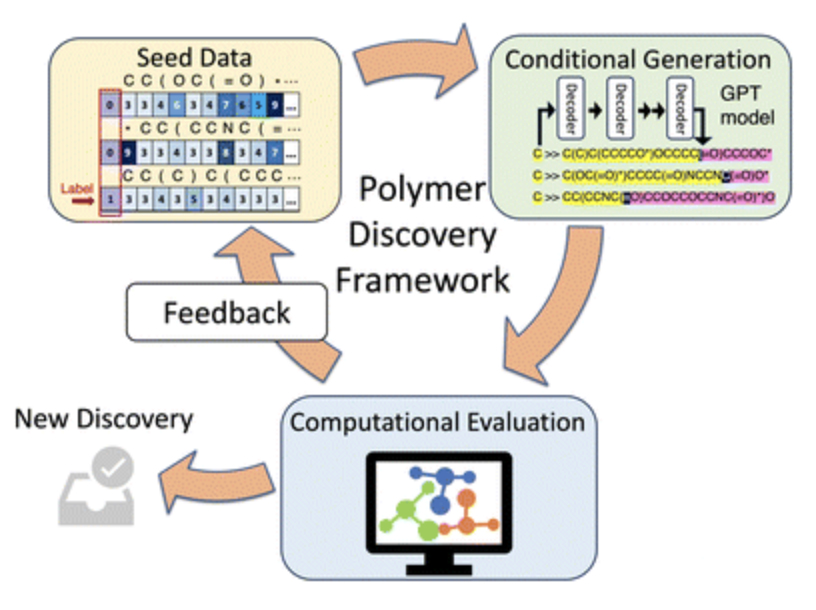
In this work, we introduce a computational polymer discovery framework that efficiently designs polymers with tailored properties. The framework comprises three core components—a conditioned generative model, a computational evaluation module, and a feedback mechanism—all integrated into an iterative framework for material innovation. To demonstrate the efficacy of this framework, we used it to design polymer electrolyte materials with high ionic conductivity. A conditional generative model based on the minGPT architecture can generate candidate polymers that exhibit a mean ionic conductivity that is greater than that of the original training set. This approach, coupled with molecular dynamics (MD) simulations for testing and a specifically planned acquisition mechanism, allows the framework to refine its output iteratively. Notably, we observe an increase in both the mean and the lower bound of the ionic conductivity of the new polymer candidates. The framework's effectiveness is underscored by its identification of 14 distinct polymer repeating units that display a computed ionic conductivity surpassing that of polyethylene oxide (PEO).
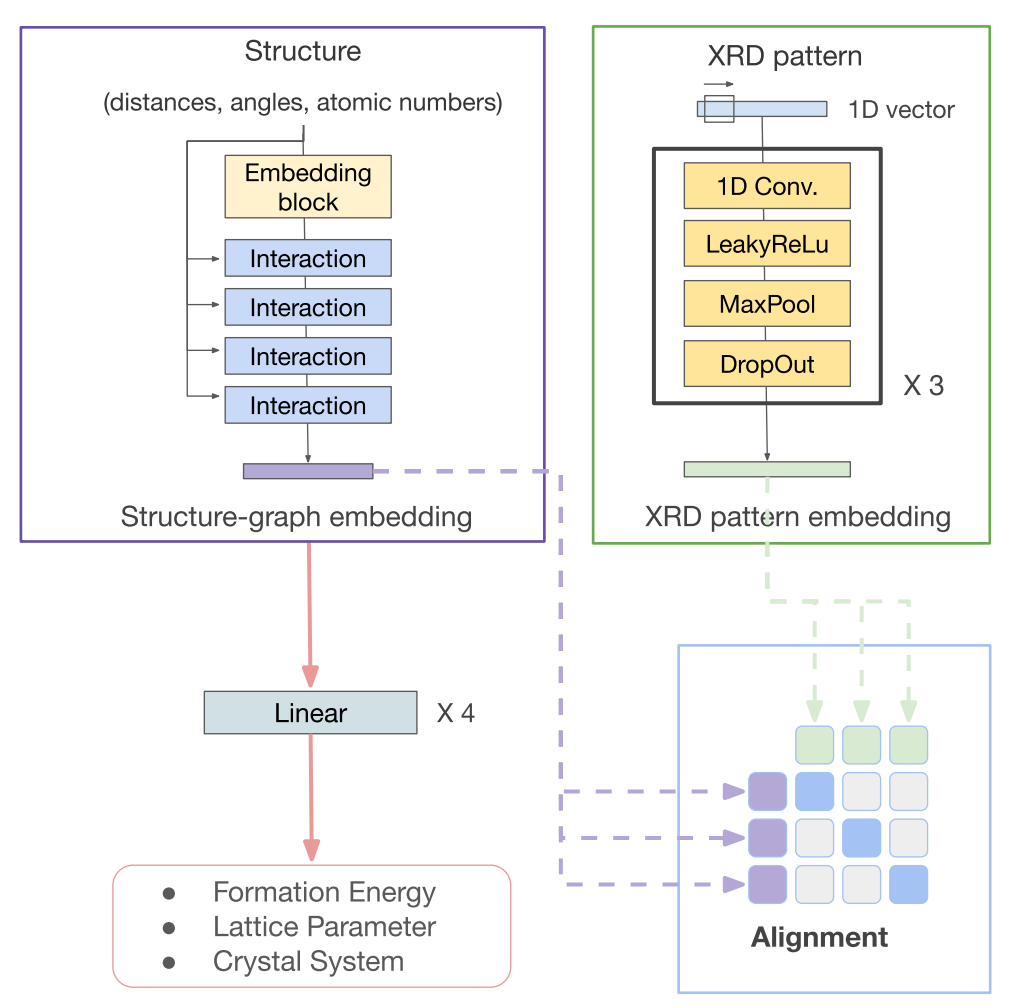
Materials science datasets are inherently heterogeneous and are available in different modalities such as characterization spectra, atomic structures, microscopic images, and text-based synthesis conditions. The advancements in multi-modal learning, particularly in vision and language models, have opened new avenues for integrating data in different forms. In this work, we evaluate common techniques in multi-modal learning (alignment and fusion) in unifying some of the most important modalities in materials science: atomic structure, X-ray diffraction patterns (XRD), and composition. We show that structure graph modality can be enhanced by aligning with XRD patterns. Additionally, we show that aligning and fusing more experimentally accessible data formats, such as XRD patterns and compositions, can create more robust joint embeddings than individual modalities across various tasks. This lays the groundwork for future studies aiming to exploit the full potential of multi-modal data in materials science, facilitating more informed decision-making in materials design and discovery. READ MORE
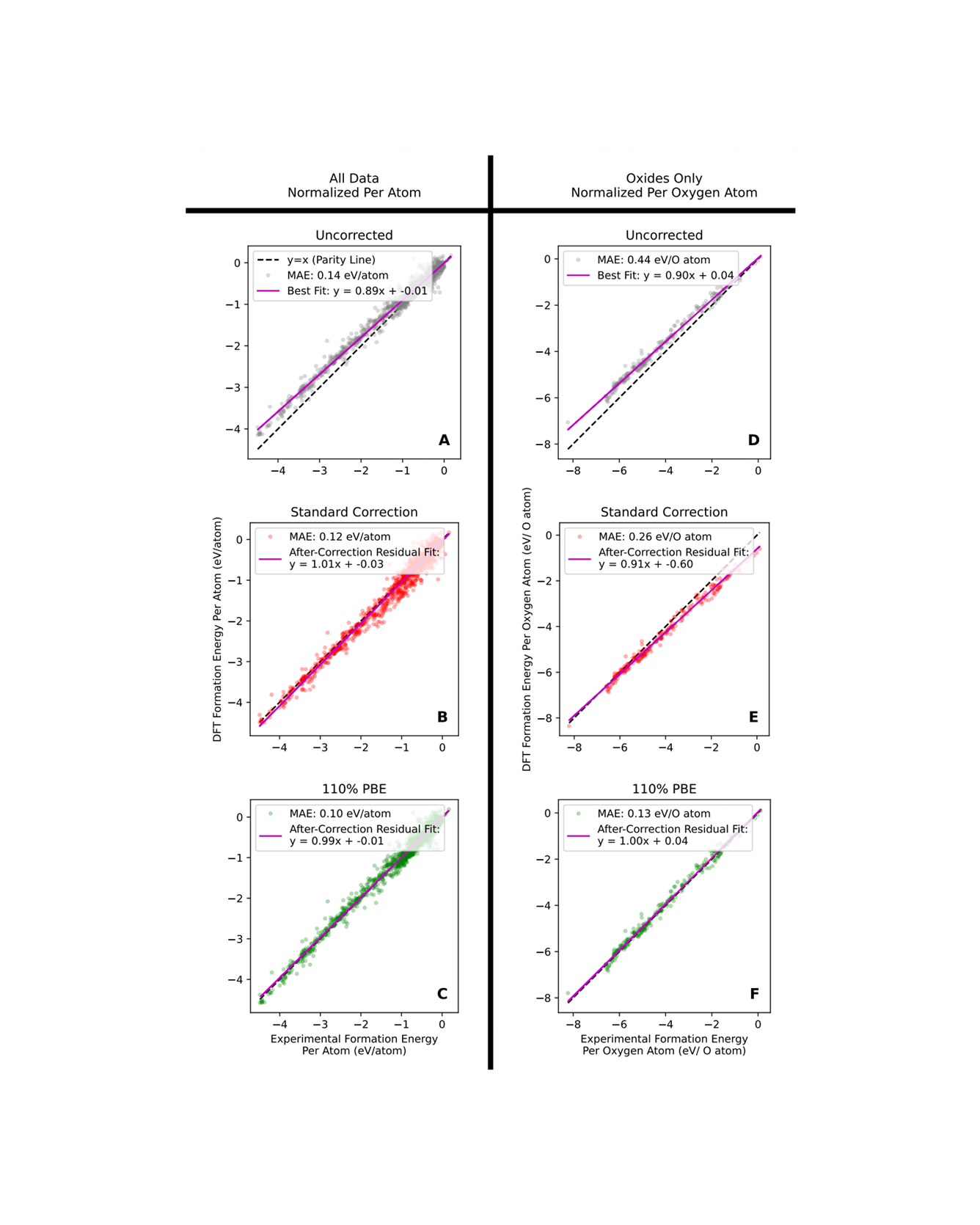
Material properties calculated using density functional theory (DFT) are often corrected to more closely match experimental values, but the most common correction method has flaws that lead to unphysical results and false positives in the material discovery process. In this work, we show that these flaws stem from the fact that only additive errors are considered, and we provide evidence that DFT predictions are likely subject to proportional error as well. We analyze the case of formation energy predictions since stability is a critical material property. We propose a simpler, linear formation energy correction method, which we call the 110% PBE correction, that models proportional error and thereby addresses the problems associated with the most common correction method. We demonstrate that the conclusions drawn from the 110% PBE correction method are more likely to be physically accurate. READ MORE
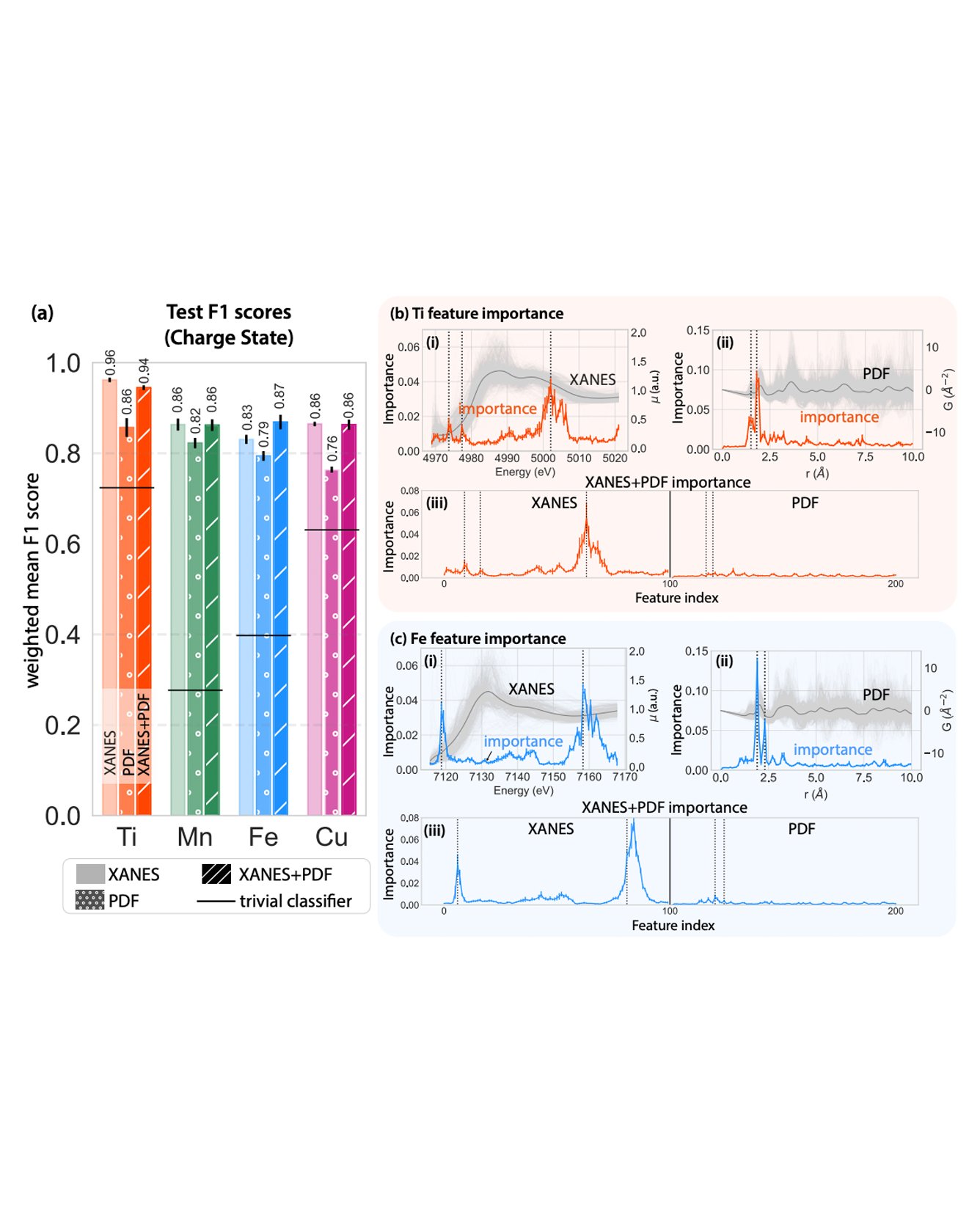
We used off-the-shelf interpretable ML techniques to combine information from multiple heterogeneous spectra: X-ray absorption near-edge spectra (XANES) and atomic pair distribution functions (PDFs), to extract information about local structure and chemistry of transition metal oxides. This approach enabled us to analyze the relative contributions of the different spectra to different prediction tasks. Specifically, we trained random forest models on XANES, PDF, and both of them combined, to extract charge (oxidation) state, coordination number, and mean nearest-neighbor bond length of transition metal cations in oxides. We find that XANES-only models tend to outperform the PDF-only models for all the tasks, and information from XANES often dominated when the two inputs were combined. This was even true for structural tasks where we might expect PDF to dominate. However, the performance gap closes when we used species-specific differential PDFs (dPDFs) as the inputs instead of total PDFs. Our results highlight that XANES contains rich structural information and may be further developed as a structural probe. Our interpretable, multimodal approach is quick and easy to implement when suitable structural and spectroscopic databases are available. This approach provides valuable insights into the relative strengths of different modalities for a practical scientific goal, guiding researchers in their experiment design tasks such as deciding when it is useful to combine complementary techniques in a scientific investigation. READ MORE
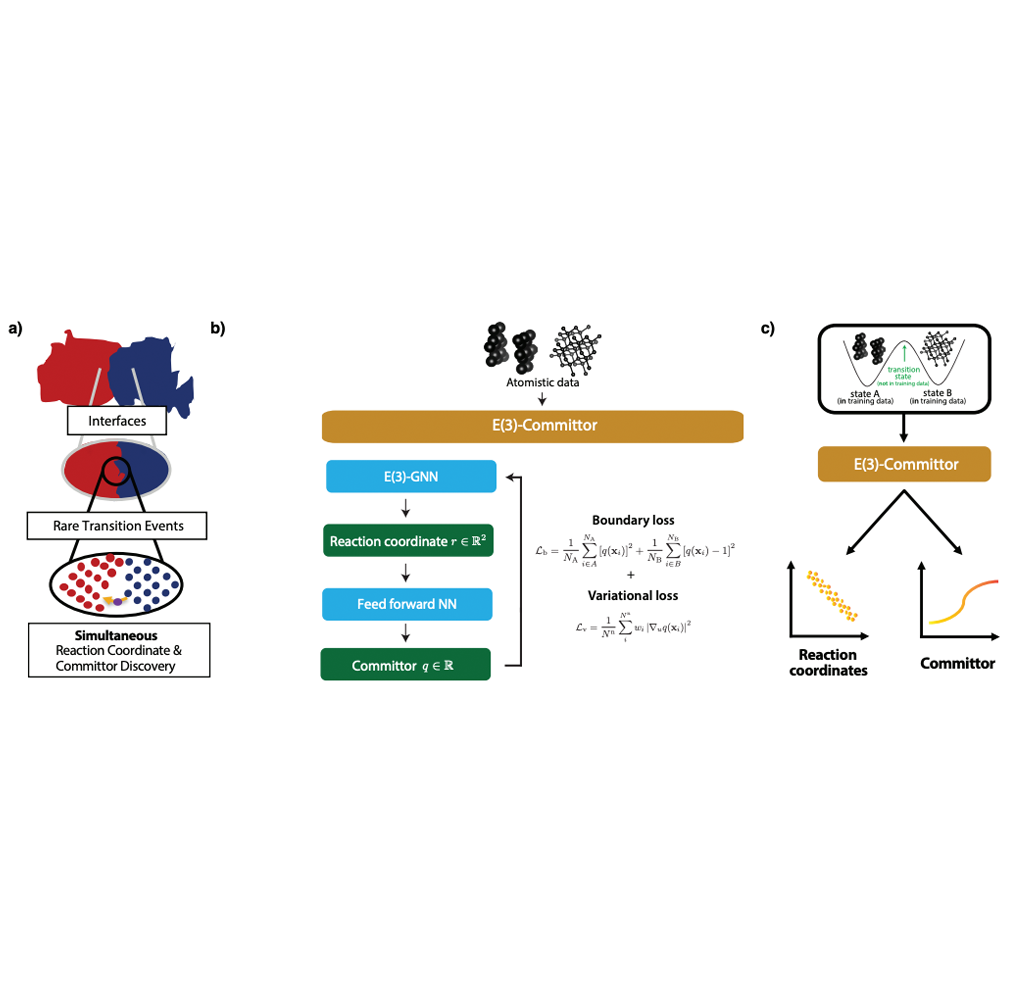
Atoms rearrange themselves during materials synthesis; understanding this self-organization choreography would help the design of novel synthesis recipes. Yet, the mechanisms of such phase transformations are often governed by statistically improbable atomic transitions - known as rare events - that are challenging to investigate by direct, brute-force sampling with conventional atomistic simulations. The transition-state theory framework has been successfully applied for numerous rare-event sampling techniques, which require prior knowledge of reaction coordinates to be encoded in a committor function. Here we show how E(3)-equivariant graph neural networks can be used to simultaneously learn physically appropriate reaction coordinates and committors, solely from molecular dynamics simulations near the start and end states of a reaction. This approach is applied to two dramatically different systems and associated mechanisms, namely the conformational transition in a alanine dipeptide molecule and the solid-liquid transition in the solidification of the CrFeNi metallic alloy. We demonstrated that this approach reduces the need for human intervention in designing reaction coordinates and committor functions, which may enable the high-throughput study of transition states. READ MORE
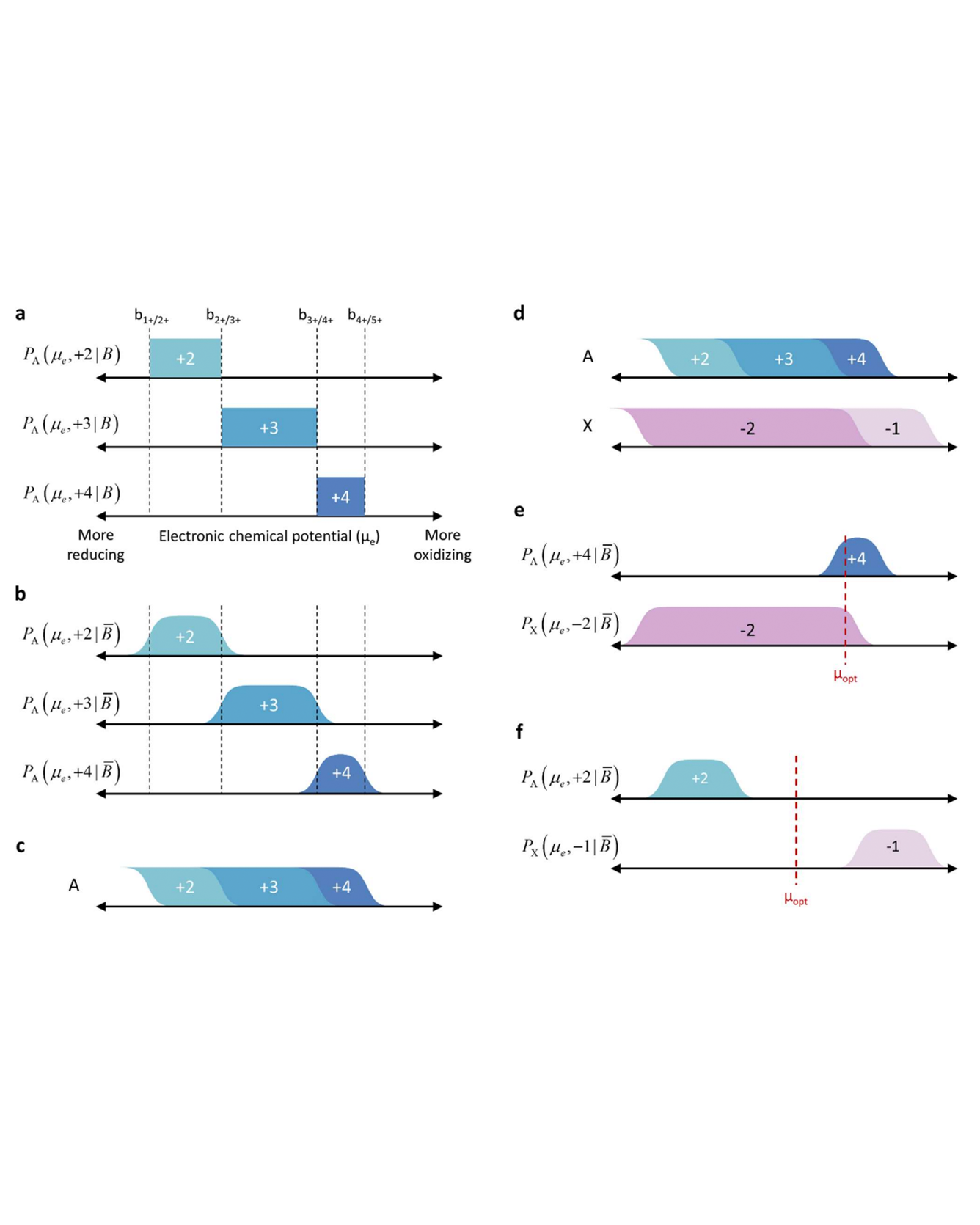
The electrochemical series is a useful tool in electrochemistry, but its effectiveness in materials chemistry is limited by the fact that the standard electrochemical series is based on a relatively small set of reactions, many of which are measured in aqueous solutions. To address this problem, we have used machine learning to create an electrochemical series for inorganic materials from tens of thousands of entries in the Inorganic Crystal Structure Database. We demonstrate that this series is generally more consistent with oxidation states in solid-state materials than the series based on aqueous ions. The electrochemical series was constructed by developing and parameterizing a physical, human-interpretable model of oxidation states in materials. We show that this model enables the prediction of oxidation states from composition in a way that is more accurate than a state-of-the-art transformer-based neural network model. We present applications of our approach to structure prediction, materials discovery, and materials electrochemistry, and we discuss possible additional applications and areas for improvement. To facilitate the use of our approach, we introduce a freely available website and API. READ MORE
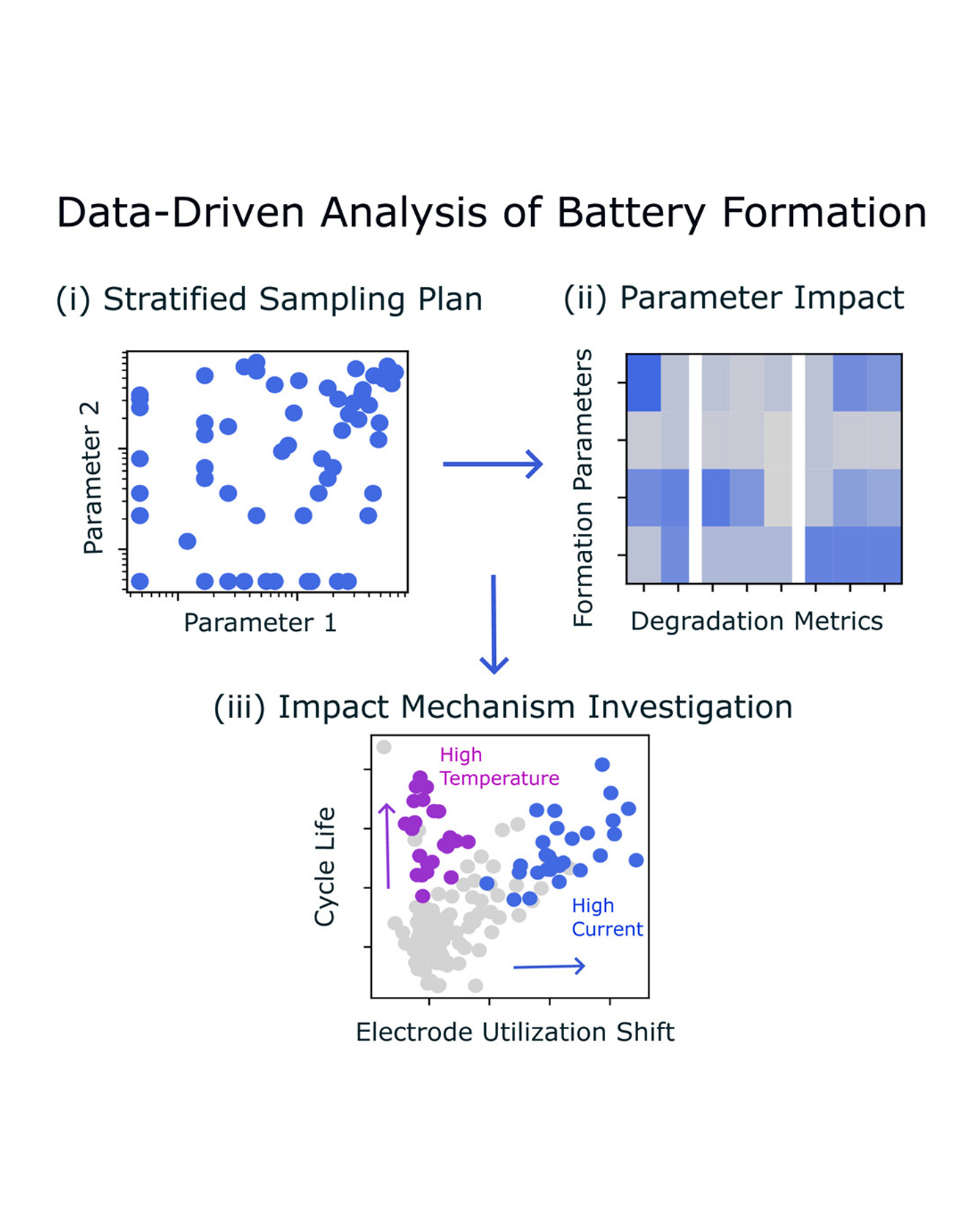
Formation is a critical step in battery manufacturing. During this process, lithium inventory is consumed to form the solid electrolyte interphase (SEI), which in turn determines the battery lifetime. To tackle the vast parameter space and complexity of formation, we employ a data-driven workflow on 186 lithium-ion battery cells across 62 formation protocols. We identify two key parameters, formation charge current and temperature, that control battery longevity via distinct mechanisms. Surprisingly, high-formation charge current on the first cycle extends battery cycle life by an average of 50%. Unlike elevated formation temperature, which boosts battery performance by forming a robust SEI, the cycle life improvement for fast-formed cells arises from a shifted electrode-specific utilization after formation. Apart from the widely acknowledged role of formation in governing SEI properties, we demonstrate how formation protocols determine the stoichiometry range over which the positive and negative electrodes are cycled. READ MORE
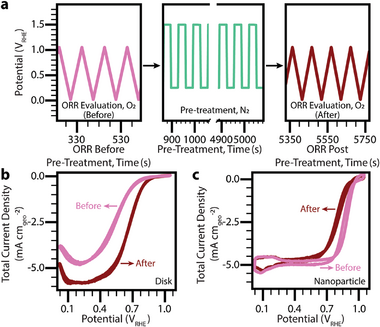
Activation, break-in, and/or pre-treatment protocols are generally applied to energy conversion devices before regular operation to reach stable performance. There remains much to understand about the relationships among physical properties, performance, and electrochemical pre-treatments. Here, a design-of-experiments (DoE) strategy is employed to address this gap by demonstrating the influence of five pre-treatment parameters for carbon-supported Pt-nanoparticle catalysts on the electrocatalytic oxygen reduction reaction (ORR). A subset of pre-treatments, developed using a central composite design, are tested in a flow cell combined with an inductively-coupled plasma mass spectrometer (on-line ICP-MS). The DoE-based approach facilitates comprehensive insights from two orders of magnitude fewer experiments than a conventional grid search. The coupled on-line ICP-MS setup enables effective catalysis and real-time catalyst dissolution data. Leveraging insights from DoE for on-line ICP-MS and additional characterization, a model is built between the degradation of a multi-dimensional supported Pt surface, its performance, and applied electrochemical parameters. These investigations identify surface modifications, such as oxidation, and subsequent restructuring of Pt during pre-treatment as a primary cause of performance deterioration during ORR. By combining DoE with advanced characterization techniques, a powerful approach is demonstrated to gain a mechanistic understanding of pre-treatment protocols that can be broadly adapted to various reaction chemistries.
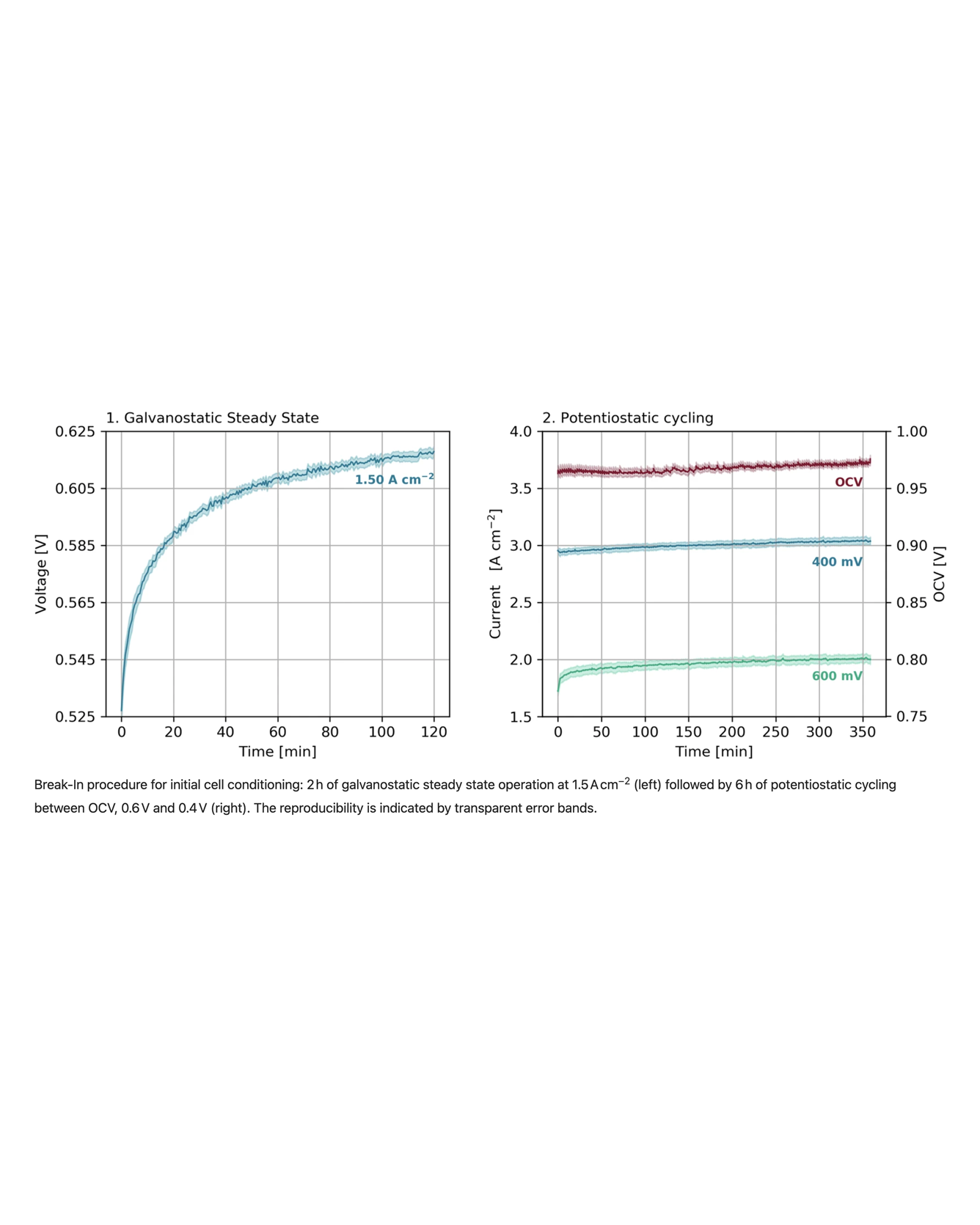
The composition and morphology of the cathode catalyst layer (CCL) have a significant impact on the performance and stability of polymer electrolyte membrane fuel cells (PEMFC). Understanding the primary degradation mechanism of the CCL and its influencing factors is crucial for optimizing PEMFC performance and durability. Within this work, we present comprehensive in-situ characterization data focused on cathode catalyst degradation. The dataset consists of 36 unique durability tests with over 4000 testing hours, including variations in the cathode ionomer to carbon ratio, platinum on carbon ratio, ionomer equivalent weight, and carbon support type. The applied accelerated stress tests were conducted with different upper potential limits and relative humidities. Characterization techniques including IV-curves, limiting current measurements, electrochemical impedance spectroscopy, and cyclic voltammetry were employed to analyse changes in performance, charge and mass transfer, and electrochemically active surface area of the catalyst. The aim of the dataset is to improve the understanding of catalyst degradation by allowing comparisons across material variations and provide practical information for other researchers in the field. READ MORE