Featured Publications
All Publications
TRI Authors: Jiexiong Tang (intern), Hanme Kim, Vitor Guizilini, Sudeep Pillai, Rares Ambrus
All Authors: Jiexiong Tang, Hanme Kim, Vitor Guizilini, Sudeep Pillai, Rares Ambrus
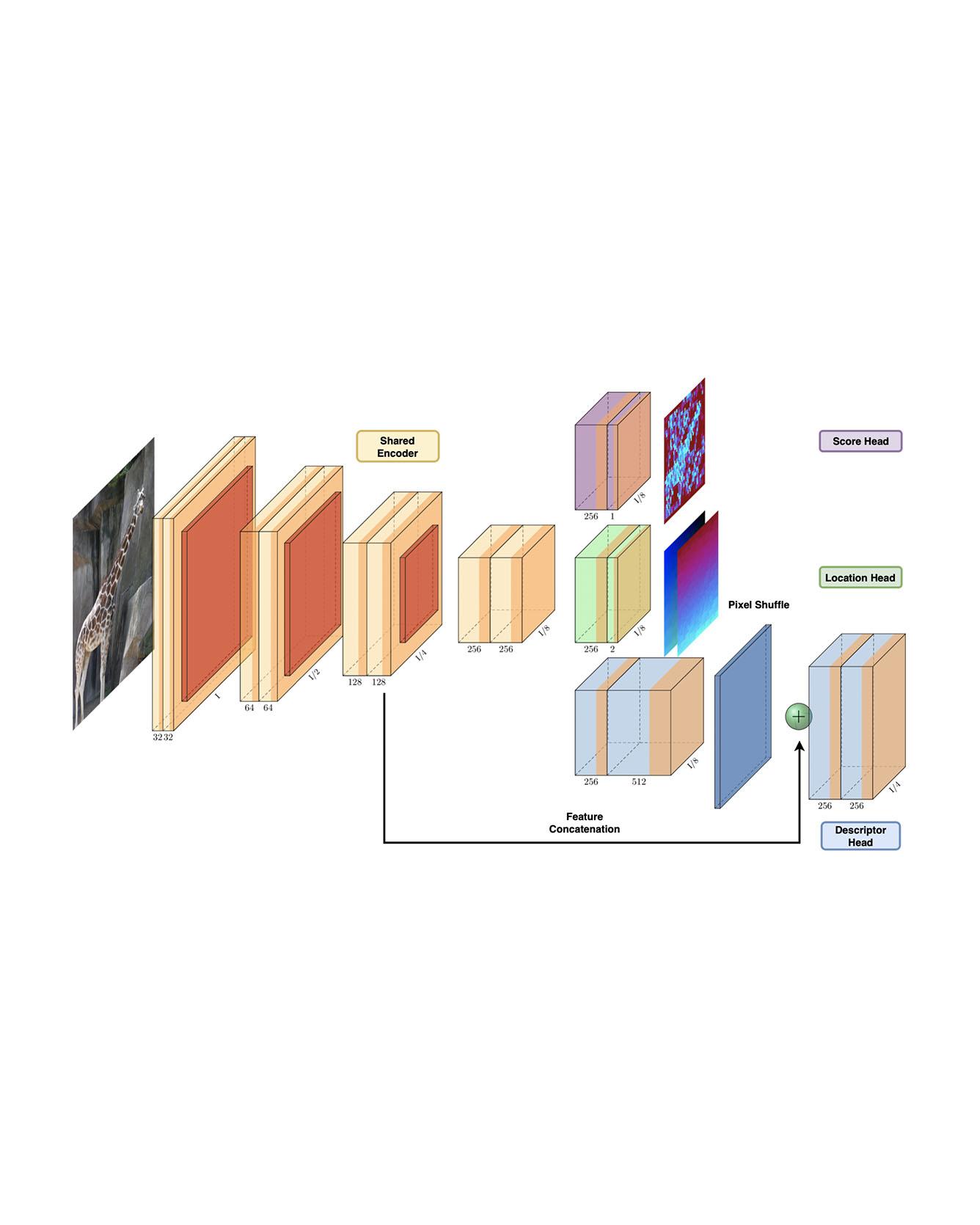
Identifying salient points in images is a crucial component for visual odometry, Structure-from-Motion or SLAM algorithms. Recently, several learned keypoint methods have demonstrated compelling performance on challenging benchmarks. However, generating consistent and accurate training data for interest-point detection in natural images still remains challenging, especially for human annotators. We introduce IO-Net (i.e. InlierOutlierNet), a novel proxy task for the self-supervision of keypoint detection, description and matching. By making the sampling of inlier-outlier sets from point-pair correspondences fully differentiable within the keypoint learning framework, we show that are able to simultaneously self-supervise keypoint description and improve keypoint matching. Second, we introduce KeyPointNet, a keypoint-network architecture that is especially amenable to robust keypoint detection and description. We design the network to allow local keypoint aggregation to avoid artifacts due to spatial discretizations commonly used for this task, and we improve fine-grained keypoint descriptor performance by taking advantage of efficient sub-pixel convolutions to upsample the descriptor feature-maps to a higher operating resolution. Through extensive experiments and ablative analysis, we show that the proposed self-supervised keypoint learning method greatly improves the quality of feature matching and homography estimation on challenging benchmarks over the state-of-the-art. Read More
Citation: Tang, Jiexiong, Hanme Kim, Vitor Guizilini, Sudeep Pillai, and Rares Ambrus. "Neural Outlier Rejection for Self-Supervised Keypoint Learning." ICLR 2020.
TRI Authors: Vitor Guizilini, Jie Li, Rares Ambrus, Adrien Gaidon
All Authors: Vitor Guizilini, Rui Hou, Jie Li, Rares Ambrus, Adrien Gaidon Self-supervised learning is showing great promise for monocular depth estimation, using geometry as the only source of supervision. Depth networks are indeed capable of learning representations that relate visual appearance to 3D properties by implicitly leveraging category-level patterns. In this work we investigate how to leverage more directly this semantic structure to guide geometric representation learning, while remaining in the self-supervised regime. Instead of using semantic labels and proxy losses in a multi-task approach, we propose a new architecture leveraging fixed pretrained semantic segmentation networks to guide self-supervised representation learning via pixel-adaptive convolutions. Furthermore, we propose a two-stage training process to overcome a common semantic bias on dynamic objects via resampling. Our method improves upon the state of the art for self-supervised monocular depth prediction over all pixels, fine-grained details, and per semantic categories. Read more
Citation: Guizilini, Vitor, Rui Hou, Jie Li, Rares Ambrus, and Adrien Gaidon. "Semantically-Guided Representation Learning for Self-Supervised Monocular Depth." ICLR 2020

TRI Authors: Kuppuswamy, Naveen*, Alex Alspach, Avinash Uttamchandani, Sam Creasey, Takuya Ikeda, Russ Tedrake.
All Authors: Kuppuswamy, Naveen*, Alex Alspach, Avinash Uttamchandani, Sam Creasey, Takuya Ikeda, Russ Tedrake.
Citation: Kuppuswamy, Naveen*, Alex Alspach, Avinash Uttamchandani, Sam Creasey, Takuya Ikeda, Russ Tedrake. "Soft-bubbles grippers for robust and perceptive manipulation." 2020 IEEE/RSJ International Conference on Intelligent Robots and Systems.

TRI Authors: Naveen Kuppuswamy, Alejandro Castro, Calder Phillips-Grafflin, Alex Alspach, Russ Tedrake
All Authors: Naveen Kuppuswamy, Alejandro Castro, Calder Phillips-Grafflin, Alex Alspach, Russ Tedrake
Modeling deformable contact is a well-known problem in soft robotics and is particularly challenging for compliant interfaces that permit large deformations. We present a model for the behavior of a highly deformable dense geometry sensor in its interaction with objects; the forward model predicts the elastic deformation of a mesh given the pose and geometry of a contacting rigid object. We use this model to develop a fast approximation to solve the inverse problem: estimating the contact patch when the sensor is deformed by arbitrary objects. This inverse model can be easily identified through experiments and is formulated as a sparse Quadratic Program (QP) that can be solved efficiently online. The proposed model serves as the first stage of a pose estimation pipeline for robot manipulation. We demonstrate the proposed inverse model through real-time estimation of contact patches on a contact-rich manipulation problem in which oversized fingers screw a nut onto a bolt, and as part of a complete pipeline for pose-estimation and tracking based on the Iterative Closest Point (ICP) algorithm. Our results demonstrate a path towards realizing soft robots with highly compliant surfaces that perform complex real-world manipulation tasks. Read More
Citation: Kuppuswamy, Naveen, Alejandro Castro, Calder Phillips-Grafflin, Alex Alspach, and Russ Tedrake. "Fast model-based contact patch and pose estimation for highly deformable dense-geometry tactile sensors." IEEE Robotics and Automation Letters (2019).

TRI Authors: Muratahan Aykol, Joseph Montoya, Jens Hummelshøj
All Authors: Roni Choudhury, Muratahan Aykol, Samuel Gratzl, Joseph Montoya, Jens Hummelshøj
Materials science research deals primarily with understanding the relationship between the structure and properties of materials. With recent advances in computational power and automation of simulation techniques, material structure and property databases have emerged (Curtarolo et al., 2012; Jain et al., 2013; Kirklin et al., 2015), allowing a more data-driven approach to carrying out materials research. Recent studies have demonstrated that representing these databases as material networks can enable extraction of new materials knowledge (Hegde, Aykol, Kirklin, & Wolverton, 2018; Isayev et al., 2015) or help tackle challenges like predictive synthesis (Aykol, Hegde, et al., 2019) that require relational information between materials. Materials databases have become very popular because they enable their users to do rapid prototyping by searching near globally for figures of merit for their target application. However, both scientists and engineers have little in the way of visualization of aggregates from these databases, that is, intuitive layouts that help understand which materials are related and how they are related. The need for a tool that does this is particularly crucial in materials science because properties like phase stability and crystal structure similarity are themselves functions of a material dataset, rather than of individual materials. Read More
Citation: Choudhury, Roni, Muratahan Aykol, Samuel Gratzl, Joseph Montoya, and Jens Hummelshøj. "MaterialNet: A web-based graph explorer for materials science data." Journal of Open Source Software 5, no. 47 (2020): 2105.

TRI Authors: Muratahan Aykol*
All Authors: Vinay Hegde, Muratahan Aykol*, Scott Kirklin, Chris Wolverton*
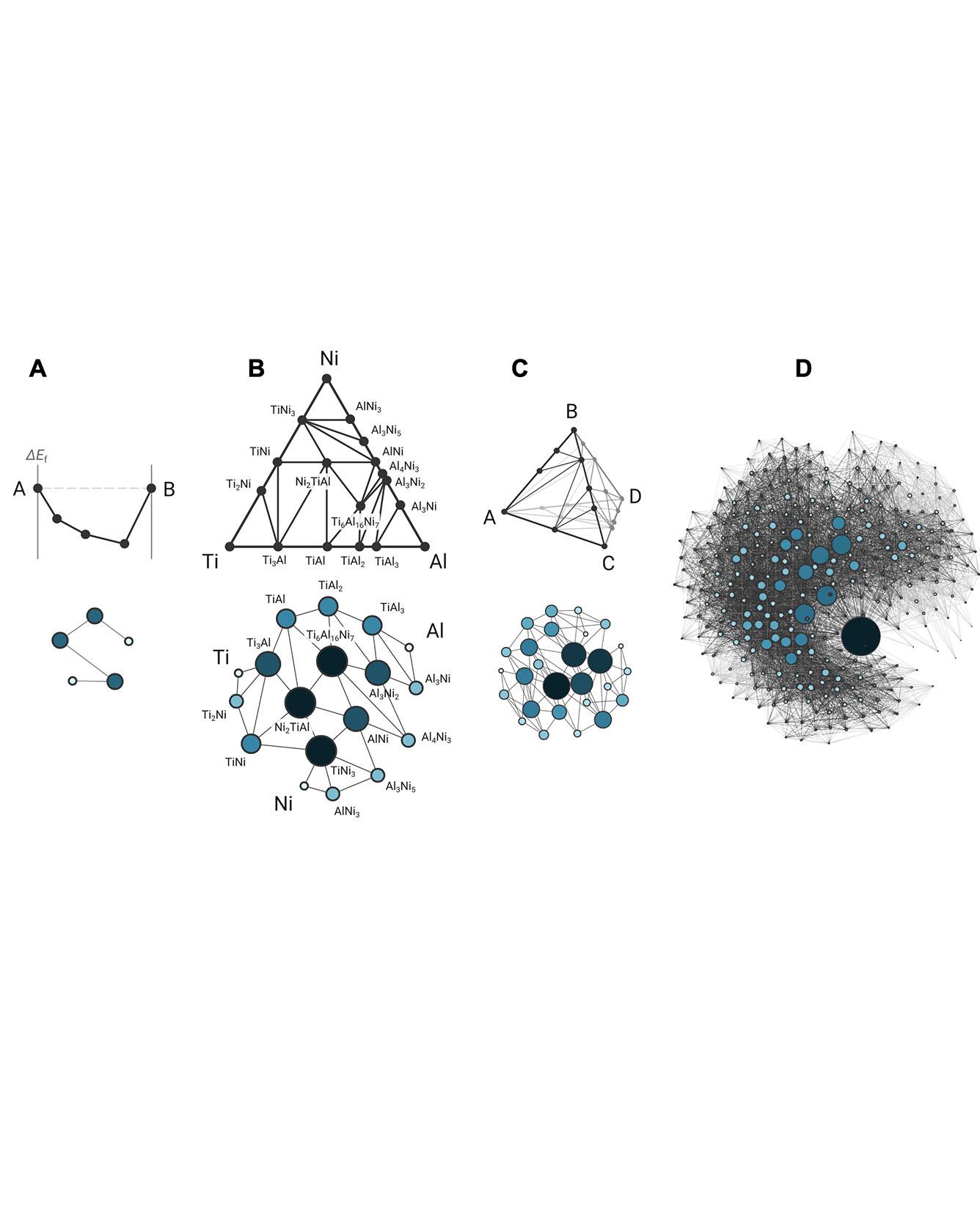
One of the holy grails of materials science, unlocking structure-property relationships, has largely been pursued via bottom-up investigations of how the arrangement of atoms and interatomic bonding in a material determine its macroscopic behavior. Here, we consider a complementary approach, a top-down study of the organizational structure of networks of materials, based on the interaction between materials themselves. We unravel the complete “phase stability network of all inorganic materials” as a densely connected complex network of 21,000 thermodynamically stable compounds (nodes) interlinked by 41 million tie line (edges) defining their two-phase equilibria, as computed by high-throughput density functional theory. Analyzing the topology of this network of materials has the potential to uncover previously unidentified characteristics inaccessible from traditional atoms-to-materials paradigms. Using the connectivity of nodes in the phase stability network, we derive a rational, data-driven metric for material reactivity, the “nobility index,” and quantitatively identify the noblest materials in nature. Read More
Citation: Hegde, Vinay I., Muratahan Aykol, Scott Kirklin, and Chris Wolverton. "The phase stability network of all inorganic materials." Science Advances 6, no. 9 (2020): eaay5606.
TRI Authors: Patrick K Herring, Muratahan Aykol
All Authors: Peter M Attia, Aditya Grover, Norman Jin, Kristen A Severson, Todor M Markov, Yang-Hung Liao, Michael H Chen, Bryan Cheong, Nicholas Perkins, Zi Yang, Patrick K Herring, Muratahan Aykol, Stephen J Harris, Richard D Braatz, Stefano Ermon, William C Chueh
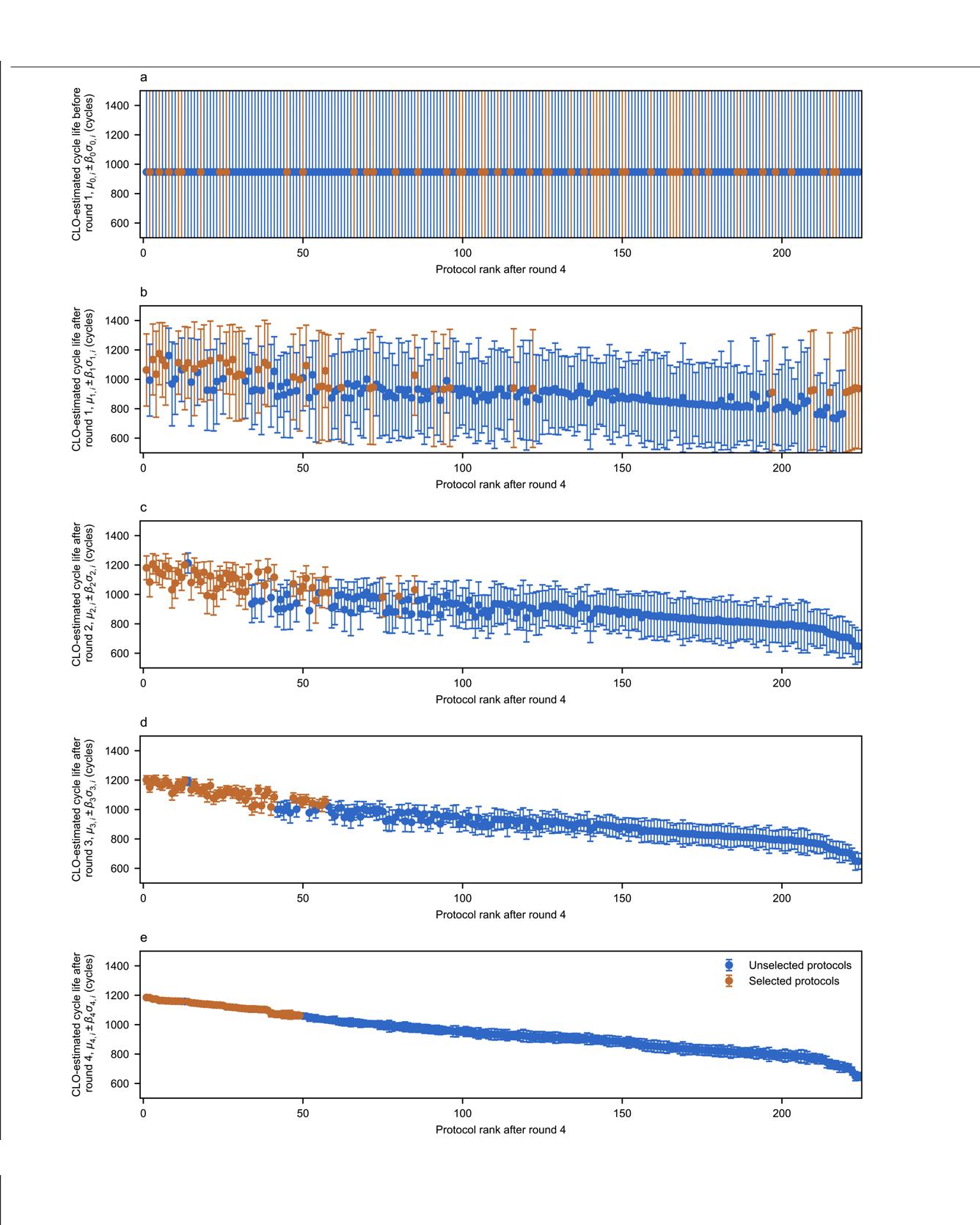
Simultaneously optimizing many design parameters in time-consuming experiments causes bottlenecks in a broad range of scientific and engineering disciplines1,2. One such example is process and control optimization for lithium-ion batteries during materials selection, cell manufacturing and operation. A typical objective is to maximize battery lifetime; however, conducting even a single experiment to evaluate lifetime can take months to years3,4,5. Furthermore, both large parameter spaces and high sampling variability3,6,7 necessitate a large number of experiments. Hence, the key challenge is to reduce both the number and the duration of the experiments required. Here we develop and demonstrate a machine learning methodology to efficiently optimize a parameter space specifying the current and voltage profiles of six-step, ten-minute fast-charging protocols for maximizing battery cycle life, which can alleviate range anxiety for electric-vehicle users8,9. We combine two key elements to reduce the optimization cost: an early-prediction model5, which reduces the time per experiment by predicting the final cycle life using data from the first few cycles, and a Bayesian optimization algorithm10,11, which reduces the number of experiments by balancing exploration and exploitation to efficiently probe the parameter space of charging protocols. Using this methodology, we rapidly identify high-cycle-life charging protocols among 224 candidates in 16 days (compared with over 500 days using exhaustive search without early prediction), and subsequently validate the accuracy and efficiency of our optimization approach. Our closed-loop methodology automatically incorporates feedback from past experiments to inform future decisions and can be generalized to other applications in battery design and, more broadly, other scientific domains that involve time-intensive experiments and multi-dimensional design spaces. Read More
Citation: Attia, Peter M., Aditya Grover, Norman Jin, Kristen A. Severson, Todor M. Markov, Yang-Hung Liao, Michael H. Chen et al. "Closed-loop optimization of fast-charging protocols for batteries with machine learning." Nature 578, no. 7795 (2020): 397-402.
TRI Authors: German Ros
All Authors: Michael R Anderson, Michael Cafarella, Thomas F Wenisch, German Ros
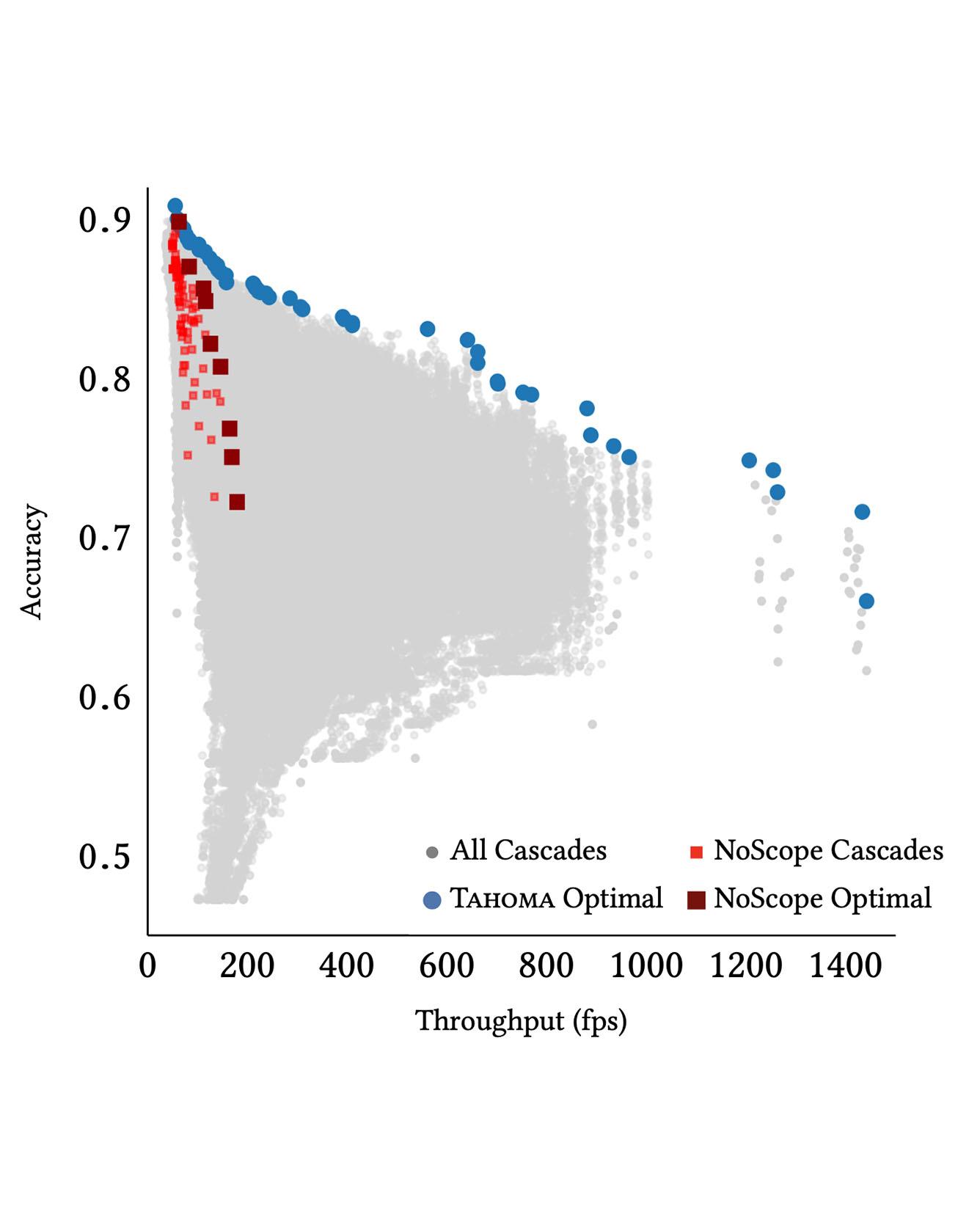
Querying the content of images, video, and other non-textual data sources requires expensive content extraction methods. Modern extraction techniques are based on deep convolutional neural networks (CNNs) and can classify objects within images with astounding accuracy. Unfortunately, these methods are slow, needing several milliseconds per image using modern GPUs. The cost of content-based queries over a huge video corpus is prohibitive. Read More
Citation: Anderson, Michael R., Michael Cafarella, Thomas F. Wenisch, and German Ros. "Predicate optimization for a visual analytics database." SysML 2018
TRI Author: Brian D. Storey
All Authors: Hongbo Zhao, Brian D. Storey, Richard D. Braatz, and Martin Z. Bazant
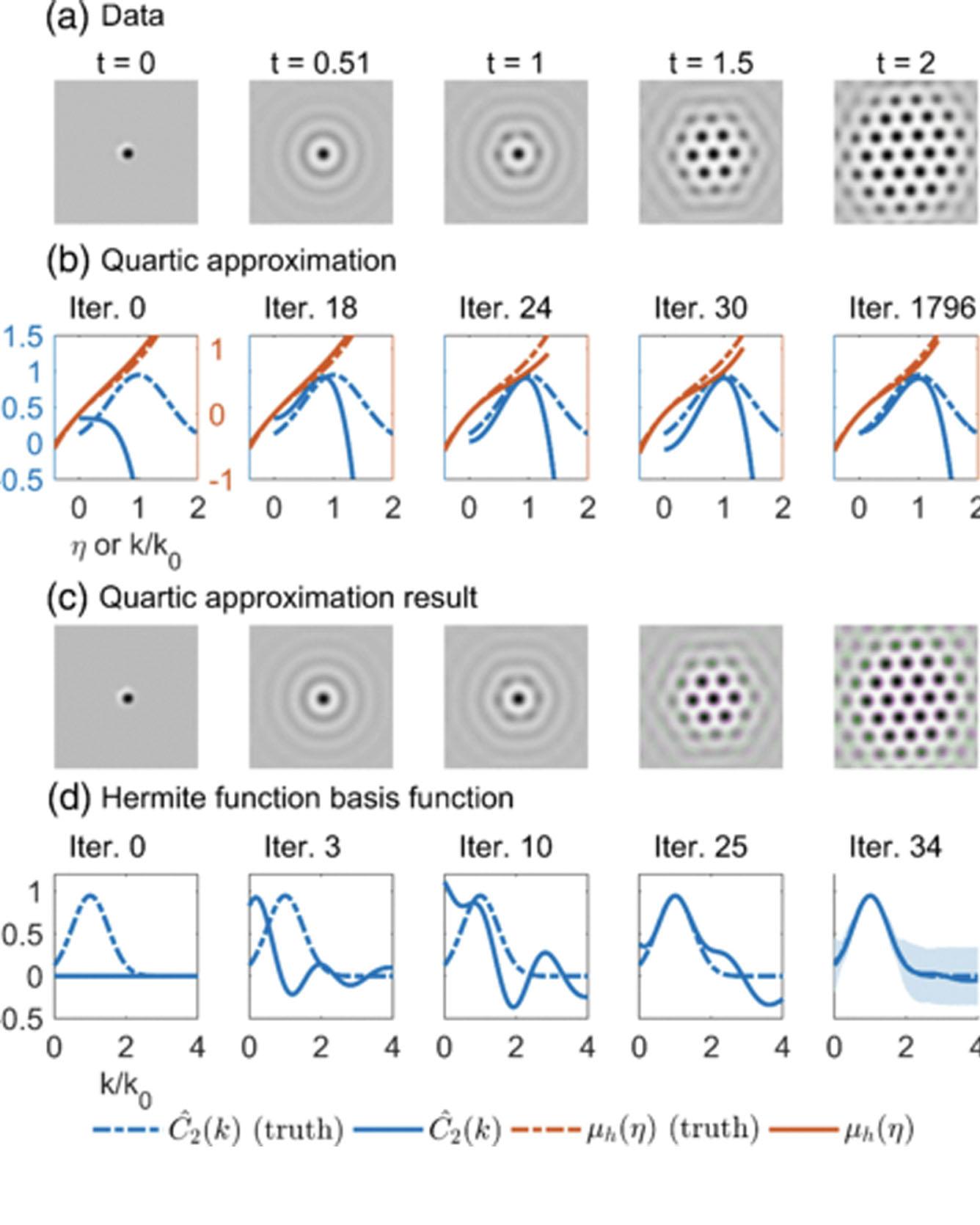
Using a framework of partial differential equation-constrained optimization, we demonstrate that multiple constitutive relations can be extracted simultaneously from a small set of images of pattern formation. Examples include state-dependent properties in phase-field models, such as the diffusivity, kinetic prefactor, free energy, and direct correlation function, given only the general form of the Cahn-Hilliard equation, Allen-Cahn equation, or dynamical density functional theory (phase-field crystal model). Constraints can be added based on physical arguments to accelerate convergence and avoid spurious results. Reconstruction of the free energy functional, which contains nonlinear dependence on the state variable and differential or convolutional operators, opens the possibility of learning nonequilibrium thermodynamics from only a few snapshots of the dynamics. Read More
Citation: Zhao, Hongbo, Brian D. Storey, Richard D. Braatz, and Martin Z. Bazant. "Learning the Physics of Pattern Formation from Images." Physical Review Letters 124, no. 6 (2020): 060201.
TRI Author: Montoya, J. H.
All Authors: Mrdjenovich, D., Horton, M. K., Montoya, J. H., Legaspi, C. M., Dwaraknath, S., Tshitoyan, V., Jain A., Persson, K. A
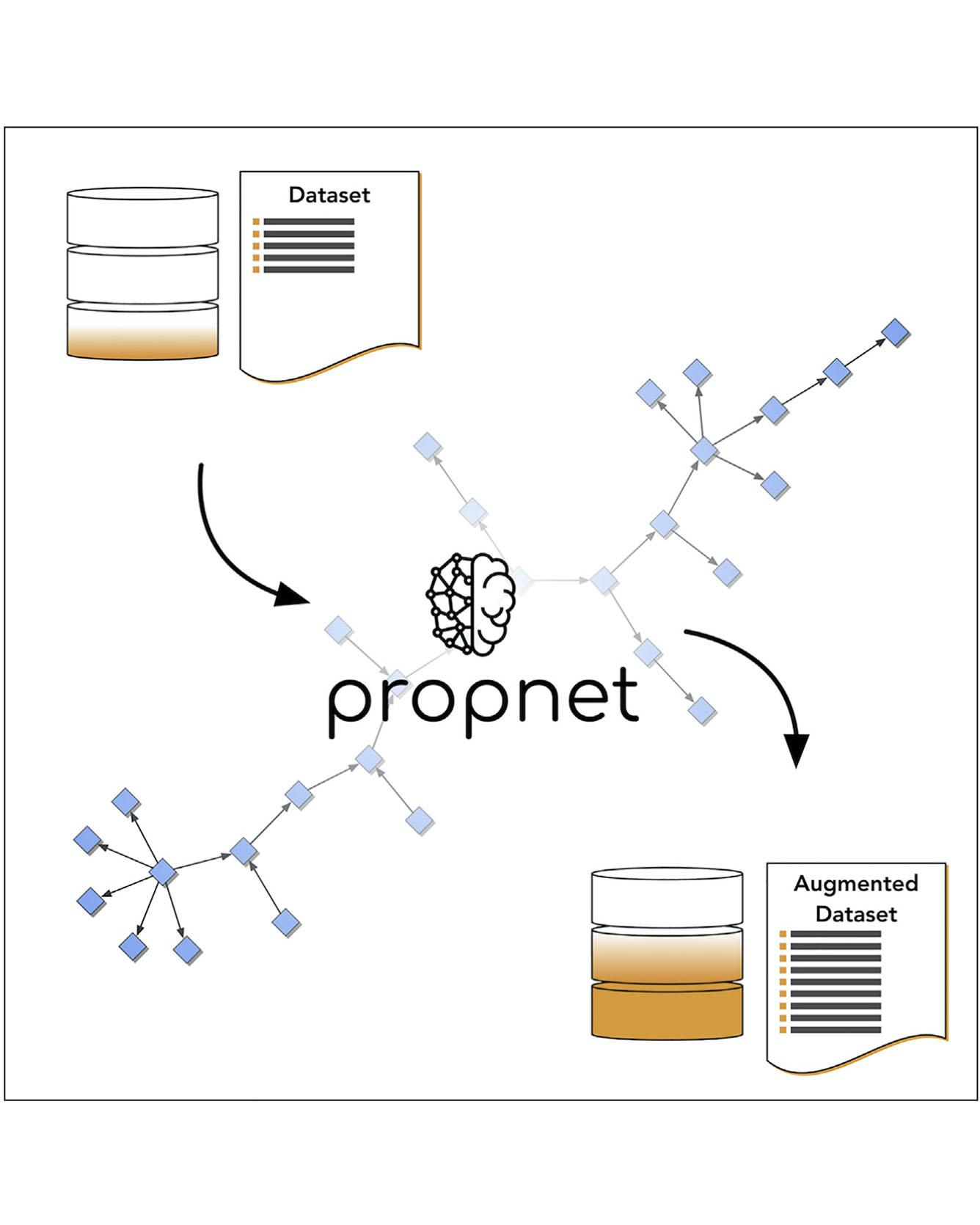
Data-driven materials science is bolstered by the recent growth of online materials databases. However, the current informatics infrastructure has yet to unlock the full knowledge available within existing datasets or to explore connections between different materials science domains. Here, we present a streamlined system for codifying and connecting materials properties in an open-source Python framework: propnet. We demonstrate the capability of this framework to augment existing datasets of materials properties: by consecutively applying a network of physical relationships to calculate related information, propnet connects disparate domain knowledge. Beyond an immediate increase in available information, the results allow for the examination of correlations between sets of properties and guide the design of multifunctional materials. By emphasizing code extensibility and simplicity, we offer this software to the materials science community for general application to any experimental or computationally derived materials database. Read More
Citations: Mrdjenovich, David, Matthew K. Horton, Joseph H. Montoya, Christian M. Legaspi, Shyam Dwaraknath, Vahe Tshitoyan, Anubhav Jain, and Kristin A. Persson. "propnet: A Knowledge Graph for Materials Science." Matter (2020).