
Featured Publications

All Publications
TRI Authors: Alejandro Castro, Ante Qu (intern), Naveen Kuppuswamy, Alex Alspach, Michael Sherman
All Authors: Alejandro Castro, Ante Qu, Naveen Kuppuswamy, Alex Alspach, Michael Sherman
Multibody simulation with frictional contact has been a challenging subject of research for the past thirty years. Rigidbody assumptions are commonly used to approximate the physics of contact, and together with Coulomb friction, lead to challengingto-solve nonlinear complementarity problems (NCP). On the other hand, robot grippers often introduce significant compliance. Compliant contact, combined with regularized friction, can be modeled entirely with ODEs, avoiding NCP solves. Unfortunately, regularized friction introduces high-frequency stiff dynamics and even implicit methods struggle with these systems, especially during slip-stick transitions. To improve the performance of implicit integration for these systems we introduce a Transition-Aware Line Search (TALS), which greatly improves the convergence of the Newton-Raphson iterations performed by implicit integrators. We find that TALS works best with semi-implicit integration, but that the explicit treatment of normal compliance can be problematic. To address this, we develop a Transition-Aware Modified SemiImplicit (TAMSI) integrator that has similar computational cost to semi-implicit methods but implicitly couples compliant contact forces, leading to a more robust method. We evaluate the robustness, accuracy and performance of TAMSI and demonstrate our approach alongside relevant sim-to-real manipulation tasks. Read More
Citation: Castro, Alejandro M., Ante Qu, Naveen Kuppuswamy, Alex Alspach, and Michael Sherman. "A Transition-Aware Method for the Simulation of Compliant Contact with Regularized Friction." IEEE Robotics and Automation Letters 5, no. 2 (2020): 1859-1866.

Battery evaluation and early prediction software package (BEEP) provides an open-source Python-based framework for the management and processing of high-throughput battery cycling data-streams. BEEPs features include file-system based organization of raw cycling data and metadata received from cell testing equipment, validation protocols that ensure the integrity of such data, parsing and structuring of data into Python-objects ready for analytics, featurization of structured cycling data to serve as input for machine-learning, and end-to-end examples that use processed data for anomaly detection and featurized data to train early-prediction models for cycle life. BEEP is developed in response to the software and expertise gap between cell-level battery testing and data-driven battery development. READ MORE
TRI Authors: Adrien Gaidon, Nikos Arechiga
All Authors: Kaidi Cao, Colin Wei, Adrien Gaidon, Nikos Arechiga, Tengyu Ma
Deep learning algorithms can fare poorly when the training dataset suffers from heavy class-imbalance but the testing criterion requires good generalization on less frequent classes. We design two novel methods to improve performance in such scenarios. First, we propose a theoretically-principled label-distribution-aware margin (LDAM) loss motivated by minimizing a margin-based generalization bound. This loss replaces the standard cross-entropy objective during training and can be applied with prior strategies for training with class-imbalance such as re-weighting or re-sampling. Second, we propose a simple, yet effective, training schedule that defers re-weighting until after the initial stage, allowing the model to learn an initial representation while avoiding some of the complications associated with re-weighting or re-sampling. We test our methods on several benchmark vision tasks including the real-world imbalanced dataset iNaturalist 2018. Our experiments show that either of these methods alone can already improve over existing techniques and their combination achieves even better performance gains. Read More
Citation: Cao, Kaidi, Colin Wei, Adrien Gaidon, Nikos Arechiga, and Tengyu Ma. "Learning imbalanced datasets with label-distribution-aware margin loss." In Advances in Neural Information Processing Systems, pp. 1565-1576. 2019.
TRI Authors: Stephen McGill, Jonathan DeCastro, Luke Fletcher, John Leonard, Guy Rosman
All Authors: Huang, Xin, Stephen McGill, Jonathan DeCastro, Luke Fletcher, John Leonard, Brian Williams, Guy Rosman
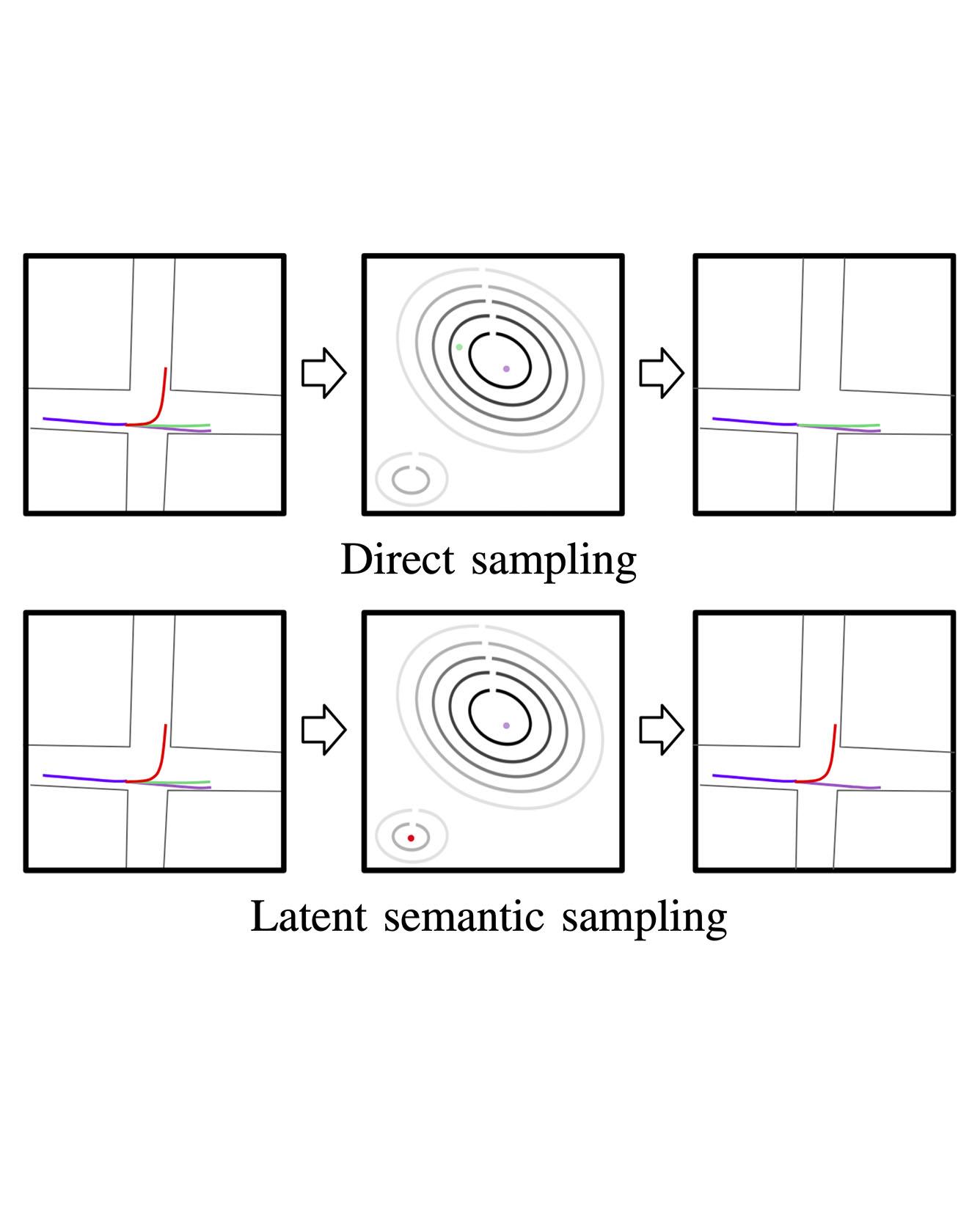
Vehicle trajectory prediction is crucial for autonomous driving and advanced driver assistant systems. While existing approaches may sample from a predicted distribution of vehicle trajectories, they lack the ability to explore it -- a key ability for evaluating safety from a planning and verification perspective. In this work, we devise a novel approach for generating realistic and diverse vehicle trajectories. We extend the generative adversarial network (GAN) framework with a low-dimensional approximate semantic space, and shape that space to capture semantics such as merging and turning. We sample from this space in a way that mimics the predicted distribution, but allows us to control coverage of semantically distinct outcomes. We validate our approach on a publicly available dataset and show results that achieve state-of-the-art prediction performance, while providing improved coverage of the space of predicted trajectory semantics. Read More
Citation: Huang, Xin, Stephen McGill, Jonathan DeCastro, Luke Fletcher, John Leonard, Brian Williams, Guy Rosman. "DiversityGAN: Diversity-Aware Vehicle Motion Prediction via Latent Semantic Sampling." Robotics and Automation Letters, with oral presentation to appear in IROS 2020.
TRI Authors: Stephen G. McGill, Guy Rosman, Luke Fletcher, John J. Leonard
All Authors: Stephen G. McGill, Guy Rosman, Teddy Ort, Alyssa Pierson, Igor Gilischenski, Brandon Araki, Luke Fletcher, Sertac Karaman, Daniela Rus, John J. Leonard
Among traffic accidents in the USA, 23% of fatal and 32% of non-fatal incidents occurred at intersections. For driver assistance systems, intersection navigation remains a difficult problem that is critically important to increasing driver safety. In this letter, we examine how to navigate an unsignalized intersection safely under occlusions and faulty perception. We propose a realtime, probabilistic, risk assessment for parallel autonomy control applications for occluded intersection scenarios. The algorithms are implemented on real hardware and are deployed in a variety of turning and merging topologies. We show phenomena that establish go/no-go decisions, augment acceleration through an intersection and encourage nudging behaviors toward intersections. Read More
Citation: McGill, Stephen, Guy Rosman, Teddy Ort, Alyssa Pierson, Igor Gilischenski, Brandon Araki, Luke Fletcher, Sertac Karaman, Daniela Rus, John J. Leonard. "Probabilistic Safety Metrics for Navigating Occluded Intersections." in International Conference on Intelligent Robots and Systems, IROS 2019, 2019

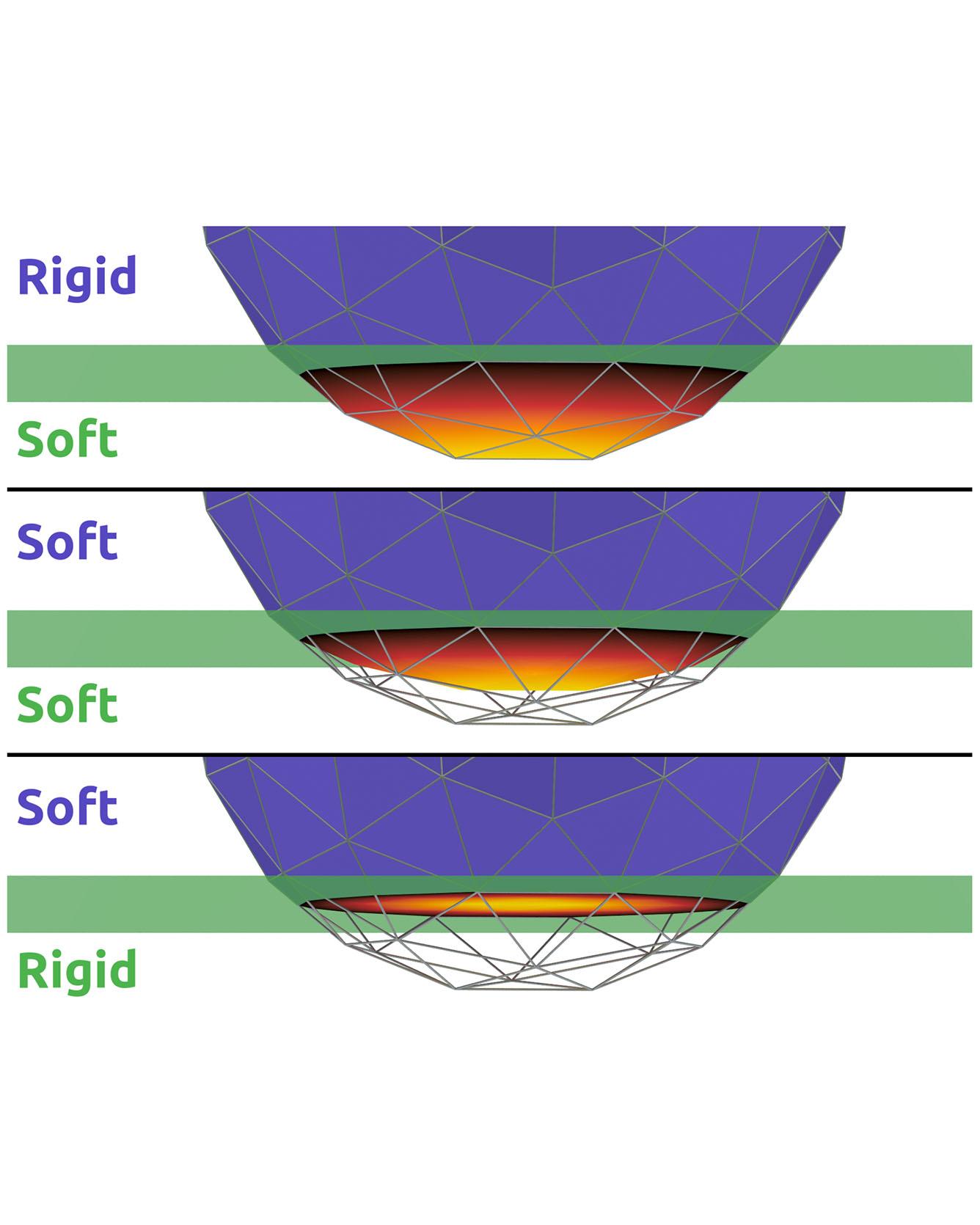
TRI Authors: Evan Drumwright, Michael Sherman All Authors: Elandt, Ryan, Evan Drumwright, Michael Sherman, and Andy Ruina We introduce an approximate model for predicting the net contact wrench between nominally rigid objects for use in simulation, control, and state estimation. The model combines and generalizes two ideas: a bed of springs (an "elastic foundation") and hydrostatic pressure. In this model, continuous pressure fields are computed offline for the interior of each nominally rigid object. Unlike hydrostatics or elastic foundations, the pressure fields need not satisfy mechanical equilibrium conditions. When two objects nominally overlap, a contact surface is defined where the two pressure fields are equal. This static pressure is supplemented with a dissipative rate-dependent pressure and friction to determine tractions on the contact surface. The contact wrench between pairs of objects is an integral of traction contributions over this surface. The model evaluates much faster than elasticity-theory models, while showing the essential trends of force, moment, and stiffness increase with contact load. It yields continuous wrenches even for non-convex objects and coarse meshes. The method shows promise as sufficiently fast, accurate, and robust for design-in-simulation of robot controllers. Read moreCitation: Elandt, Ryan, Evan Drumwright, Michael Sherman, and Andy Ruina. "A pressure field model for fast, robust approximation of net contact force and moment between nominally rigid objects." IROS 2019 arXiv preprint arXiv:1904.11433 (2019).
TRI Author: Hongkai Dai
All Authors: Bernardo Aceituno-Cabezas, Hongkai Dai, Alberto Rodriguez
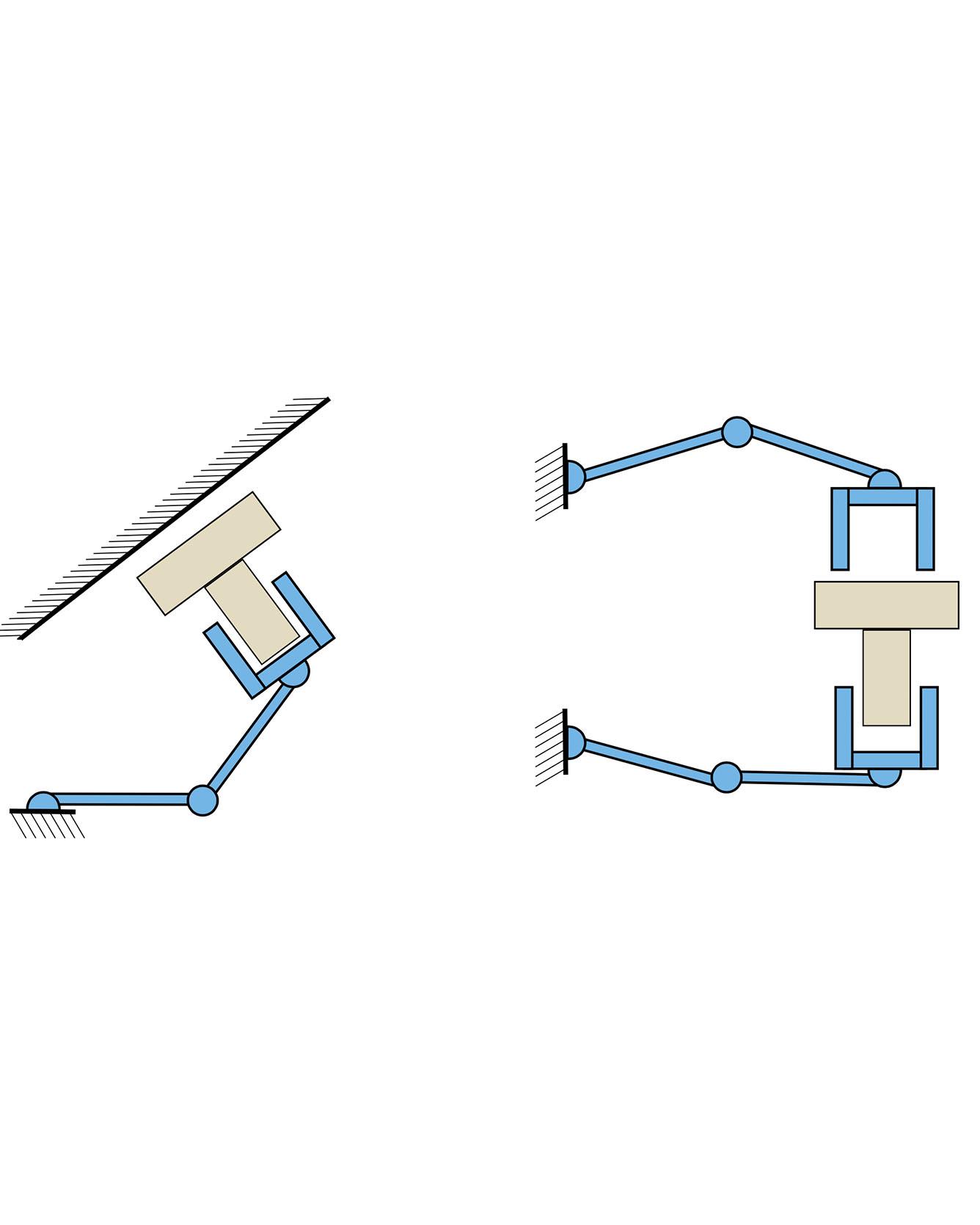
Caging is a promising tool which allows a robot to manipulate an object without directly reasoning about the contact dynamics involved. Furthermore, caging also provides useful guarantees in terms of robustness to uncertainty, and often serves as a way-point to a grasp. Unfortunately, previous work on caging is often based on computational geometry or discrete topology tools, causing restriction on gripper geometry, and difficulty on integration into larger manipulation frameworks. In this paper, we develop a convex-combinatorial model to characterize caging from an optimization perspective. More specifically, we study the configuration space of the object, where the fingers act as obstacles that enclose the configuration of the object. The convex-combinatorial nature of this approach provides guarantees on optimality, convergence and scalability, and its optimization nature makes it adaptable for further applications on robot manipulation tasks. Read More
Citation: Aceituno-Cabezas, Bernardo, Hongkai Dai, and Alberto Rodriguez. "A Convex-Combinatorial Model for Planar Caging." arXiv preprint arXiv:1809.06427 (2018).
TRI Authors: Rares Ambrus, Vitor Guizilini, Jie Li, Sudeep Pillai, Adrien Gaidon
All Authors: Rares Ambrus, Vitor Guizilini, Jie Li, Sudeep Pillai, Adrien Gaidon
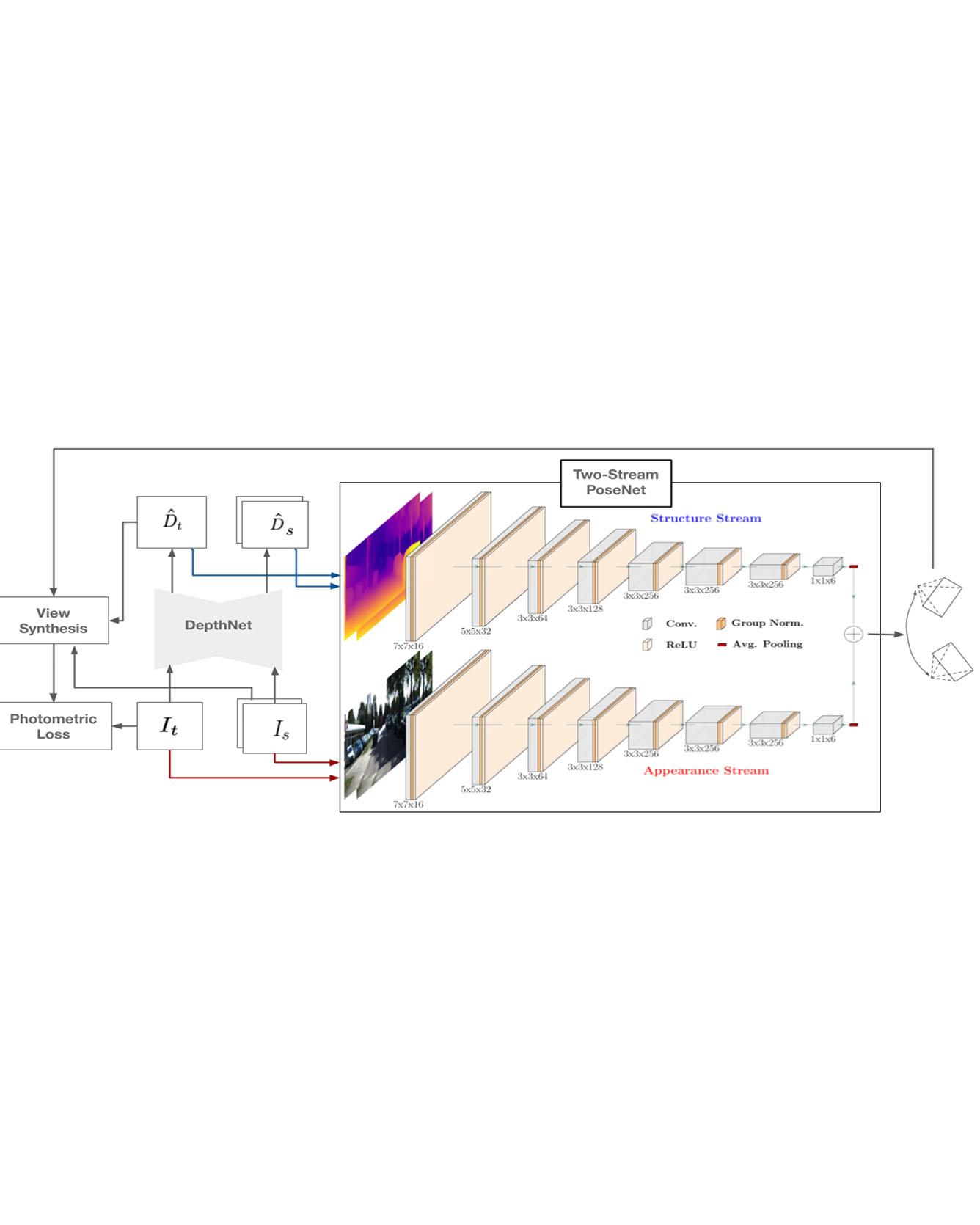
Learning depth and camera ego-motion from raw unlabeled RGB video streams is seeing exciting progress through self-supervision from strong geometric cues. To leverage not only appearance but also scene geometry, we propose a novel self-supervised two-stream network using RGB and inferred depth information for accurate visual odometry. In addition, we introduce a sparsity-inducing data augmentation policy for ego-motion learning that effectively regularizes the pose network to enable stronger generalization performance. As a result, we show that our proposed two-stream pose network achieves state-of-the-art results among learning-based methods on the KITTI odometry benchmark, and is especially suited for self-supervision at scale. Our experiments on a large-scale urban driving dataset of 1 million frames indicate that the performance of our proposed architecture does indeed scale progressively with more data. Read more
Citation: Ambrus, Rares, Vitor Guizilini, Jie Li, Sudeep Pillai, and Adrien Gaidon. "Two stream networks for self-supervised ego-motion estimation." In Conference on Robot Learning (CoRL) 2019.
TRI Authors: Vitor Guizilini, Rares Ambrus, Sudeep Pillai, Jie Li and Adrien Gaidon
All Authors: Vitor Guizilini, Rares Ambrus, Sudeep Pillai, Jie Li and Adrien Gaidon
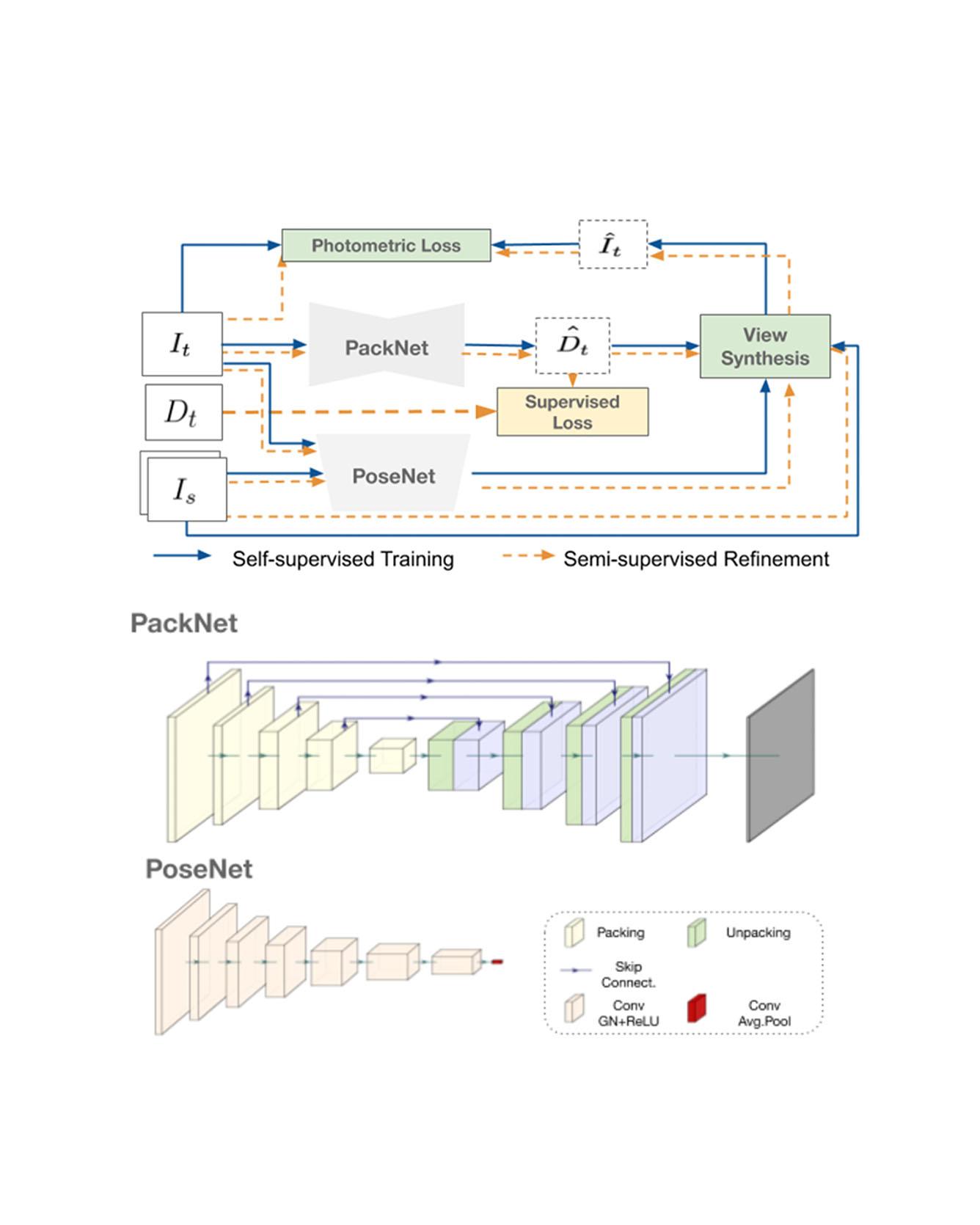
Dense depth estimation from a single image is a key problem in computer vision, with exciting applications in a multitude of robotic tasks. Initially viewed as a direct regression problem, requiring annotated labels as supervision at training time, in the past few years a substantial amount of work has been done in self-supervised depth training based on strong geometric cues, both from stereo cameras and more recently from monocular video sequences. In this paper we investigate how these two approaches (supervised & self-supervised) can be effectively combined, so that a depth model can learn to encode true scale from sparse supervision while achieving high fidelity local accuracy by leveraging geometric cues. To this end, we propose a novel supervised loss term that complements the widely used photometric loss, and show how it can be used to train robust semi-supervised monocular depth estimation models. Furthermore, we evaluate how much supervision is actually necessary to train accurate scale-aware monocular depth models, showing that with our proposed framework, very sparse LiDAR information, with as few as 4 beams (less than 100 valid depth values per image), is enough to achieve results competitive with the current state-of-the-art. Read More
Citation: Guizilini, Vitor, Jie Li, Rares Ambrus, Sudeep Pillai, and Adrien Gaidon. "Robust Semi-Supervised Monocular Depth Estimation with Reprojected Distances." In Conference on Robot Learning (CoRL) 2019.
TRI Authors: Simon Stent
All Authors: Petr Kellnhofer, Adrià Recasens, Simon Stent, Wojciech Matusik and Antonio Torralba
Understanding where people are looking is an informative social cue. In this work, we present Gaze360, a large-scale remote gaze-tracking dataset and method for robust 3D gaze estimation in unconstrained images. Our dataset consists of 238 subjects in indoor and outdoor environments with labelled 3D gaze across a wide range of head poses and distances. It is the largest publicly available dataset of its kind by both subject and variety, made possible by a simple and efficient collection method. Our proposed 3D gaze model extends existing models to include temporal information and to directly output an estimate of gaze uncertainty. We demonstrate the benefits of our model via an ablation study, and show its generalization performance via a cross-dataset evaluation against other recent gaze benchmark datasets. We furthermore propose a simple self-supervised approach to improve cross-dataset domain adaptation. Finally, we demonstrate an application of our model for estimating customer attention in a supermarket setting. Our dataset and models will be made available at http://gaze360.csail.mit.edu. Read More
Citation: Kellnhofer, Petr, Adria Recasens, Simon Stent, Wojciech Matusik, and Antonio Torralba. "Gaze360: Physically unconstrained gaze estimation in the wild." In Proceedings of the IEEE International Conference on Computer Vision, pp. 6912-6921. 2019.
